
Иммунологический аспект хронического воспаления: связь с метаболической функцией
Аннотация | Воспаление — часть врожденного иммунного ответа организма и важный процесс, который не только защищает от вредных бактерий и патогенов, но также играет ключевую роль в поддержании и восстановлении тканей. При патологических состояниях образуется двусторонняя взаимосвязь между иммунной регуляцией и аберрантным обменом веществ, что приводит к стойкому воспалению при отсутствии инфекции. Это явление называется стерильным метаболическим воспалением (метавоспалением) и возникает, если не устраняется инициирующий раздражитель или нарушается процесс разрешения. Нарушение этого жестко регулируемого иммунного ответа и его неспособность разрешиться, что, очевидно, происходит при метаболических нарушениях, не только связано с прогрессированием заболевания, но также приводит к иммунному старению и не должно игнорироваться при клиническом ведении пациентов. В этом обзоре рассмотрены механизмы, лежащие в основе хронического метаболического воспаления, аберрантной метаболической активации клеток врожденного иммунитета (нейтрофилов, макрофагов, тучных клеток, дендритных клеток) и ее роли в прогрессировании заболевания на примере ожирения–диабета. Устранение основного субклинического метаболического воспаления в дополнение к достижению контроля уровня глюкозы может внести значительный вклад в терапевтические воздействия, направленные на предотвращение возникновения сопутствующих заболеваний у пациентов с диабетом.
Введение
Воспаление представляет собой часть врожденного иммунного ответа организма и важный процесс, который не только защищает от вредных бактерий и патогенов, но также играет ключевую роль в поддержании и восстановлении тканей. Он опосредован механизмами, в которых участвуют иммунные клетки (лейкоциты), питательное микроокружение, доступность субстрата, а также секреторные факторы (белки острой фазы, протеазы, цитокины/хемокины и факторы комплемента) (Gasteiger et al. 2017, Kurita et al. 2021). При патологических состояниях существует двусторонняя перекрестная связь между иммунной регуляцией и аберрантным метаболизмом, что приводит к стойкому воспалению при отсутствии инфекции (Furman et al. 2019). Это явление называется стерильным метаболическим воспалением (метавоспаление); оно возникает, если инициирующий раздражитель не устранен или нарушен процесс разрешения (рис. 1 и 2). Таким хроническим метаболическим воспалением низкой степени не следует пренебрегать, поскольку оно в значительной степени связано с общей смертностью среди населения (Fest et al. 2019), отрицательно влияет на чувствительность к инсулину (Blaszczak et al. 2020) и повышает риск развития злокачественных новообразований (Li et al. 2023).
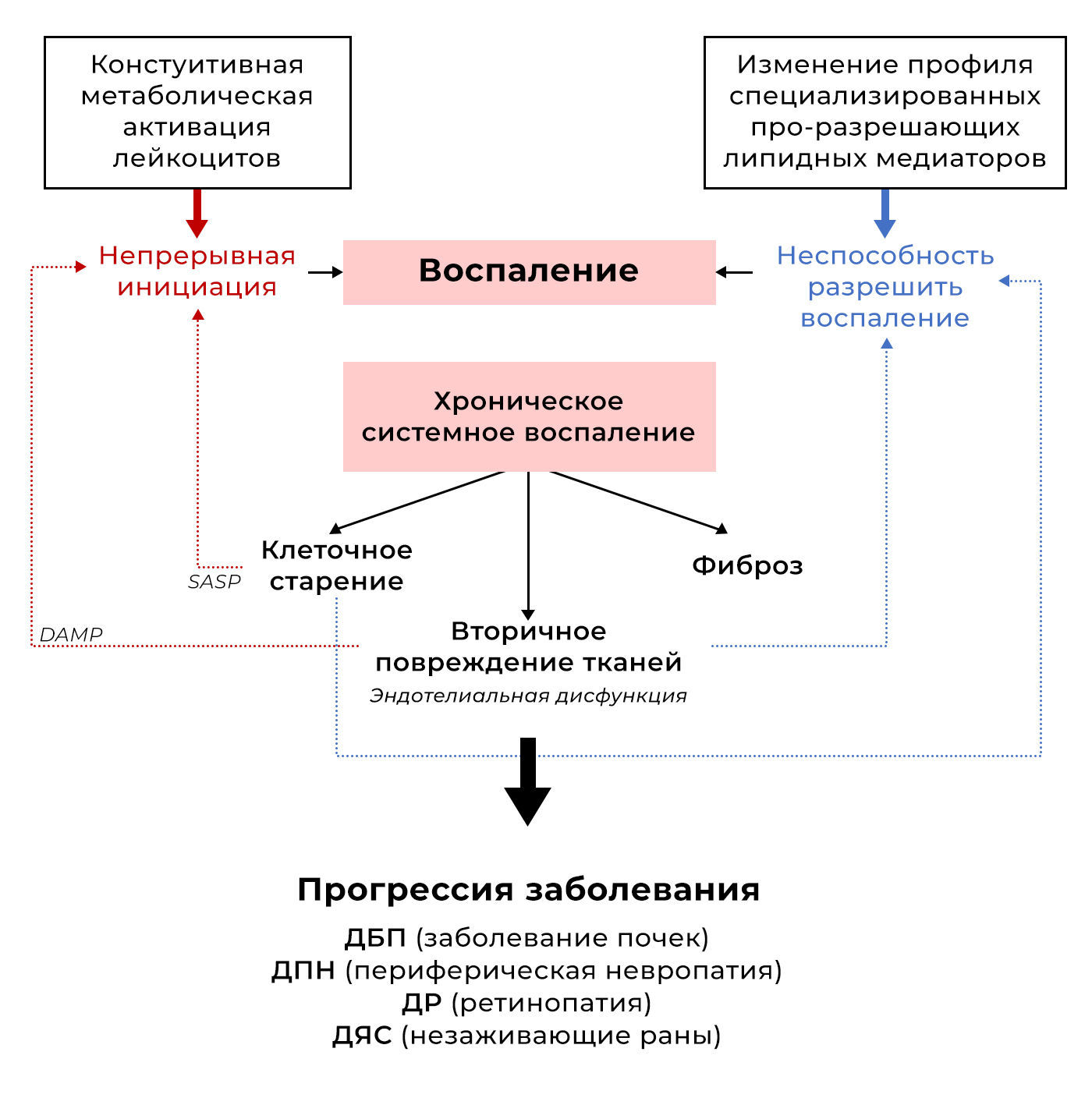
Острый воспалительный процесс хорошо описан в литературе и состоит из двух последовательных фаз, первая из которых включает нейтрофилы, привлечение моноцитов/макрофагов и их активацию, как показано на рис. 3A. В отсутствие инфекции основной целью этого начального ответа является фагоцитоз клеточного/тканевого дебриса для подготовки поврежденной/апоптотической ткани к регенерации. Сигналы опасности во внеклеточном пространстве, выделяющиеся из цитоплазмы лизирующихся клеток — молекулярные паттерны, связанные с повреждением (DAMP, также известные как алармины) — распознаются резидентными и инфильтрирующими лейкоцитами через рецепторы распознавания паттернов (PRR). PRR включают Toll-подобные рецепторы (TLR), Nod-подобные рецепторы, рецепторы комплемента, внутриклеточные рецепторы, чувствительные к нуклеиновым кислотам, и рецепторы лектина C-типа, каждый из которых запускает специфические эффекторные функции в лейкоцитах (Gasteiger et al. 2017, Chen et al. 2018, Furman et al. 2019). Во время этого начального ответа происходит сложное взаимодействие между клетками быстродействующей системы врожденного иммунитета (нейтрофилы, моноциты/макрофаги, инвариантные клетки типа естественных киллеров [iNKT], супрессорные клетки миелоидного происхождения, лимфоидные клетки врожденного иммунитета [ILC], базофилы, эозинофилы и тучные клетки) и адаптивной иммунной системы (лимфоциты, включая Th1/17, Th2, Т-регуляторные клетки [Treg] и В-клетки) (Gasteiger et al. 2017, SantaCruz-Calvo et al. 2022).
Гранулоциты, такие как нейтрофилы, являются первичными цитотоксическими эффекторными клетками, которые реагируют на воспалительные сигналы. Их активность предшествует привлечению к местам воспаления фагоцитирующих моноцитов/макрофагов, дендритных клеток и нескольких подтипов лимфоцитов (Gasteiger et al. 2017, Furman et al. 2019). Рекрутирование этих иммунных клеток хемокинами опосредуется соответствующими хемокиновыми рецепторами, связанными с G-белком (помещаемыми в соответствии с их первым цистеиновым остатком в категории C, C-C, CXC, CX3C) (Xue et al. 2019), в то время как провоспалительные цитокины для усиления воспаления действуют через гликопротеиновые рецепторы клеточной поверхности для передачи паракринных сигналов через путь янус-киназы (JAK): преобразователь сигналов и активатор транскрипции (STAT) и ядерный фактор каппа B (NF-кB) (O’Shea & Murray 2008). Цитотоксические эффекторные функции фагоцитирующих лейкоцитов вызывают истощение питательных веществ, увеличивают потребление кислорода, генерируют большое количество активных форм азота и кислорода (АФК) в тканях и могут вызывать вторичное повреждение тканей (Kominsky et al. 2010). Таким образом, за провоспалительным ответом следует фаза разрешения, во время которой нейтрофилы подвергаются регулируемой клеточной гибели (апоптозу), в то время как фагоцитирующие макрофаги переключаются на противовоспалительный фенотип, чтобы способствовать восстановлению и ремоделированию тканей. Нарушение такого жестко регулируемого иммунного ответа и неспособность его устранить, что, очевидно, происходит при метаболических нарушениях, не только связано с прогрессированием заболевания, но также приводит к иммунному старению (Thomas et al. 2021). Это особенно заметно у пациентов с ожирением и диабетом второго типа (СД2), которые, несмотря на постоянное воспаление, имеют повышенную восприимчивость к инфекциям и развитию опухолей из-за иммунного истощения (Shruthi et al. 2022).
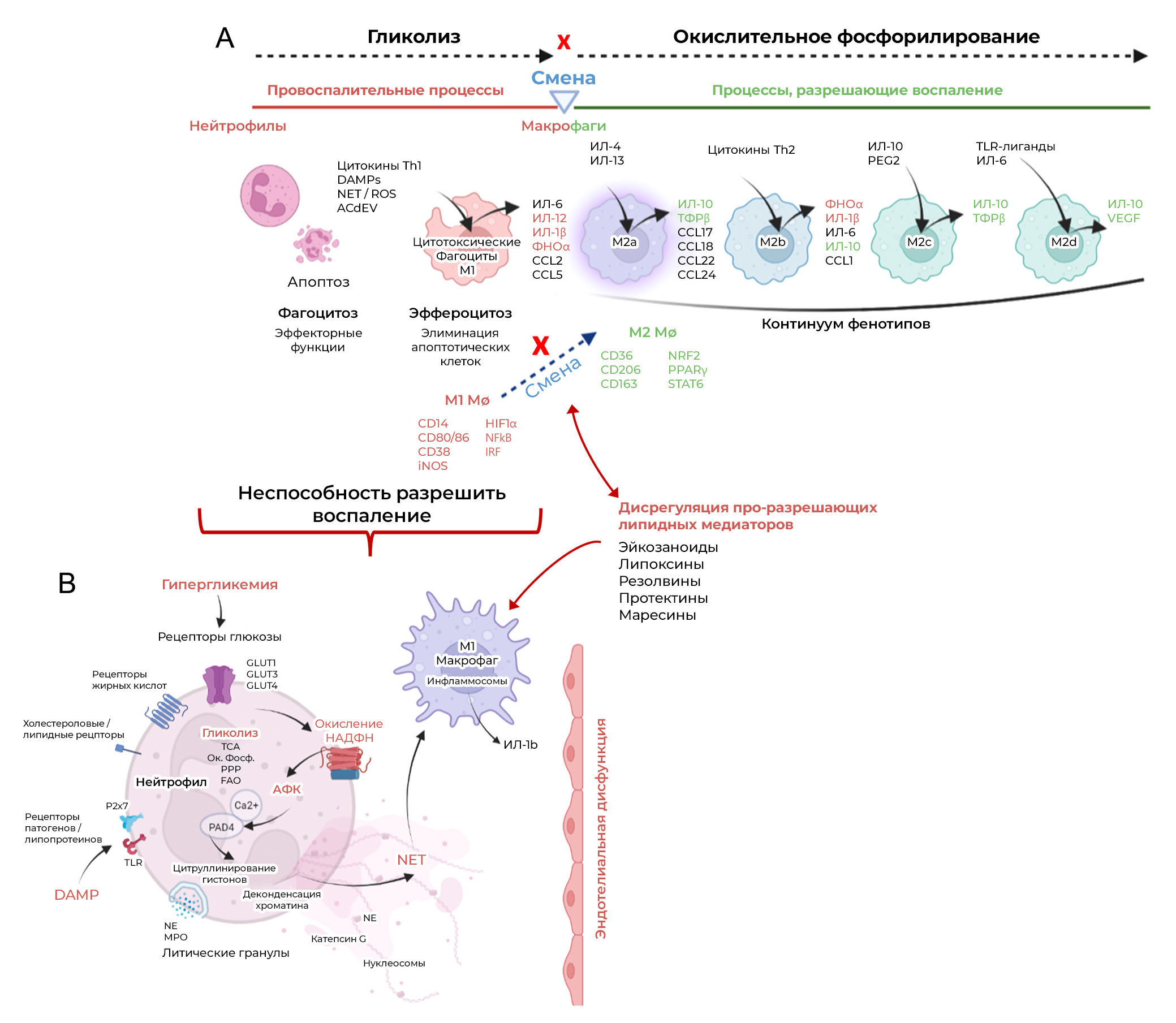
(Б) Иллюстрация конститутивной активности нейтрофилов в условиях гипергликемии, которая приводит к чрезмерному образованию ВКЛ с помощью НАДФН-оксидазы и последующей активации макрофагов M1. Неспособность макрофагов М1 переключиться на противовоспалительный фенотип связана с нарушением регуляции нескольких специализированных молекул, разрешающих воспаление (МРВ).
Воспалительные биомаркеры, такие как системный иммунно-воспалительный индекс и отношение нейтрофилов к лимфоцитам (ОНЛ), могут использоваться в качестве ранних индикаторов или предикторов клинических исходов для различных состояний, ассоциированных с диабетом, включая нефропатию (микроальбуминурию) (Chollangi et al. 2022, Qin et al. 2022), депрессию (Wang et al. 2021), макулярный отек/ретинопатию (Wan et al. 2020, Elbeyli et al. 2022), кетоацидоз (Cheng et al. 2021), язвы стопы (Vatankhah et al. 2017, Serban et al. 2021), нежелательные явления, связанные с сердечно-сосудистыми заболеваниями (Saylik & Akbulut 2022), и исход беременности при гестационном диабете (ГСД) (Wang et al. 2020, Pace & Vassallo 2021). Таким образом, устранение основного субклинического метаболического воспаления в дополнение к достижению контроля уровня глюкозы может внести значительный вклад в терапевтические вмешательства, направленные на предотвращение возникновения сопутствующих заболеваний у пациентов с диабетом.
Механизмы, лежащие в основе хронического метаболического воспаления
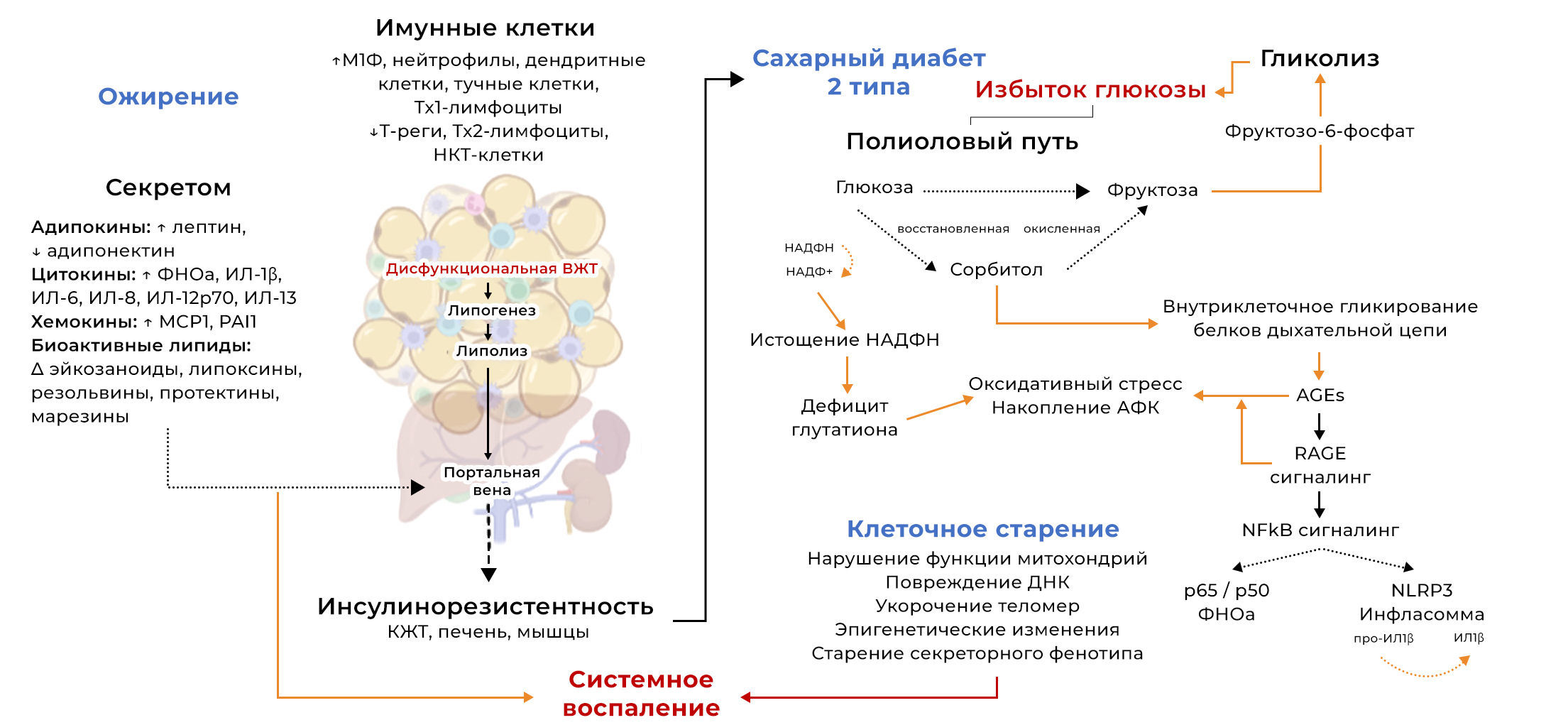
Разрастание жировой ткани и последующая гипергликемия, вызванная резистентностью к инсулину, считаются ключевыми факторами стерильного метаболического воспаления у пациентов с СД2. Связь между дисфункцией висцеральной жировой ткани (ВЖТ) и инсулинорезистентностью хорошо известна (Calderón-DuPont et al. 2022, Kahn et al. 2022). Нарушение регуляции накопления ВЖТ и ее гипертрофия при ожирении вызывают тканевую гипоксию, изменяют секреторный профиль адипокинов, цитокинов/хемокинов и биоактивных липидов, а также распределение и количество иммунных клеток в ткани (рис. 2). Животные модели дали существенное представление об изменениях, которые происходят в профиле иммунных клеток в ВЖТ после ожирения, вызванного диетой с высоким содержанием жиров. С использованием этой модели в нескольких исследованиях удалось наблюдать значительную инфильтрацию иммунных клеток, которые способствуют воспалению (нейтрофилы, фагоцитирующие макрофаги, лимфоциты Th1, дендритные клетки и тучные клетки), жировой ткани мышей с ожирением по сравнению со здоровым контролем, в то время как количество регуляторных иммунных клеток, которые уменьшают воспаление (Tregs, Th2, iNKT), снижалось (McLaughlin et al. 2017, Blaszczak et al. 2020). Известно, что разрыв гипертрофированных адипоцитов, чрезмерный липолиз и высвобождение провоспалительных медиаторов из дисфункциональной жировой ткани привлекают фагоцитирующие макрофаги с формированием короноподобных структур, окружающих поврежденные/разорванные адипоциты (Murano et al. 2008, Gasteiger et al. 2017, Gugliucci 2022). Тем не менее, роль накопления лимфоцитов в дисфункциональной жировой ткани определена менее четко, но считается, что это происходит в результате изменения секреторного профиля ВЖТ.
Обширный анализ секретома эксплантатов жировой ткани (подкожной [ПЖT] в сравнении с висцеральной [VAT]) от пациентов с ожирением подтвердил, что дисфункциональный висцеральный жир высвобождает избыточное количество провоспалительных цитокинов, адипокинов и простаноидов, как показано на рис. 2 (Kahn et al. 2022). Такой искаженный секретом не только опосредует местную инфильтрацию иммунных клеток, но также выделяется в кровоток через воротную вену, что приводит к системному воспалению и оказывает последующее функциональное влияние на чувствительность к инсулину различных других тканей, включая ПЖТ, печень и скелетные мышцы (Kahn et al. 2022, Ren et al. 2022). В исследовании ex vivo, проведенном Kahn с соавт. (2022), авторы продемонстрировали, что если факторы, выделяемые эксплантатами дисфункционального висцерального жира, воздействуют либо на гепатоциты, либо на миотубулы скелетных мышц, это отрицательно влияет на их чувствительность к инсулину, и нарушается обмен глюкозы. Таким образом, в кровотоке происходит накопление внеклеточной глюкозы (гипергликемия), в то время как доступность внутриклеточной глюкозы для синтеза глицерина и жирных кислот снижается из-за резистентности к инсулину. Это приводит к дальнейшему подавлению липогенеза (этерификации и синтеза сигнальных биоактивных липидов) в жировой ткани, в то время как нарушение антилиполитического действия инсулина приводит к увеличению циркулирующих свободных жирных кислот (Calderón-DuPont et al. 2022). В совокупности это приводит к глюкозо- и липотоксичности, что вызывет множество осложнений, связанных с мультисистемным клеточным старением и органной недостаточностью.
Длительная гипергликемия заставляет глюкозу образовывать ковалентные связи с различными белками плазмы (альбумином, фибриногеном, глобулинами, коллагеном), внутриклеточными липидами и нуклеиновыми кислотами с образованием различных типов конечных продуктов гликирования (КПГ), которые препятствуют нормальному функционированию этих молекул (Singh et al. 2014). Гликирование белков нарушает их молекулярную конформацию, изменяет активность ферментов и препятствует функционированию рецепторов. Внеклеточные КПГ связываются с рецептором конечных продуктов усиленного гликирования (RAGE) на плазматических мембранах и изменяют внутриклеточные сигнальные пути и экспрессию генов, усиливая воспаление посредством сигнального пути NF-kB (Tóbon-Velasco et al. 2014, Ramasamy et al. 2016). Известно, что эти рецепторы экспрессируются на Т-лимфоцитах, моноцитах и макрофагах и играют ключевую роль в активации иммунного ответа (Yan et al. 2008).
Кроме того, метаболический сдвиг во многих типах клеток и тканей приводит к тому, что избыток глюкозы поступает в полиоловый путь, при котором глюкоза восстанавливается до промежуточного продукта — сорбита — под действием альдозоредуктазы в НАДФH-зависимой реакции. Затем сорбитол окисляется до фруктозы с участием сорбитолдегидрогеназы. После фосфорилирования фруктоза может повторно вступить в путь гликолиза в виде фруктозо-6-фосфата. Нарушение баланса при диабете и перегрузка полиолового пути приводит к истощению НАДФН и накоплению сорбита, что, в свою очередь, вызывает обширное гликирование белков дыхательной цепи и запускает внутриклеточное повреждение, вызывая окислительный стресс и стресс эндоплазматического ретикулума за счет усиления производства цитозольных АФК (Yan 2018). Истощение НАДPH приводит к дефициту глутатиона и недостаточной антиоксидантной буферной способности для противодействия этому явлению, вызывающему нарушение функции митохондрий, повреждение ДНК и укорочение теломер (Kumar et al. 2022, Sekhar 2022). Иммунные клетки особенно чувствительны к этим патологическим изменениям и стареют, оставаясь при этом метаболически активными и приобретая ассоциированный со старением секреторный фенотип. Таким образом, в патологической микросреде, связанной с ожирением и диабетом, происходит непрерывный выброс провоспалительных сигналов различного происхождения, что постоянно усиливает воспалительную реакцию.
Точные механизмы, лежащие в основе неспособности разрешить воспаление при ожирении и диабете, еще полностью не изучены, но считается, что они связаны с многоуровневым взаимодействием между иммунной системой и метаболическими процессами, которые приводят к аберрантной активации лейкоцитов. Сюда входит (I) влияние патологического микроокружения на иммунно-метаболические реакции; (II) влияние собственного метаболизма лейкоцитов на их эффекторные функции; и (III) влияние лейкоцитов на метаболические процессы в тканях. В центре внимания этого обзора лежит нарушение регуляции клеток врожденного иммунитета в условиях метаболического воспаления; роль клеток адаптивного иммунитета рассматривается в других работах (SantaCruz-Calvo et al. 2022).
Нарушение регуляции клеток врожденного иммунитета
Нейтрофилы
Нейтрофилы (также известные как полиморфноядерные лейкоциты) считаются основными реагирующими на DAMP эффекторными клетками при стерильном метаболическом воспалении. Они образуются в костном мозге в процессе гранулопоэза, созревают и мобилизуются в кровоток с участием хемотаксических сигналов и проникают в различные ткани посредством диапедеза, где проявляют свои эффекторные функции и подвергаются апоптозу. Эффекторные функции этих лейкоцитов включают (I) окислительные взрывы и выделение литических ферментов (дегрануляция), таких как миелопероксидаза и нейтрофильная эластаза (NE), (II) фагоцитоз и продукцию провоспалительных цитокинов, и (III) образование нейтрофильных внеклеточных ловушек (НВЛ). Нейтрофилы, обладая фенотипической гетерогенностью, функционально универсальны и служат важными медиаторами воспалительной реакции (Rosales 2018). При активации нейтрофила после интегрин-зависимой адгезии к внеклеточному матриксу внутри ткани происходит быстрое образование и выделение реактивных промежуточных соединений кислорода, которое опосредуются комплексом NADPH-оксидазы (Nox) (Graham et al. 2007). Однако функциональная активность нейтрофилов зависит от поддержания внутриклеточной энергетики, и нарушения метаболизма клетки могут изменить их эффекторные функции (Kumar & Dikshit 2019, Sadiku et al. 2021). Нейтрофилы имеют очень небольшое количество митохондрий по сравнению с другими клетками и поэтому в основном для производства энергии полагаются на гликолиз (Maianski et al. 2004).
Тем не менее, нейтрофилы могут выживать в поврежденных тканях с ограниченным доступом кислорода и метаболических субстратов, полагаясь на внутриклеточные запасы гликогена и динамическую регуляцию различных других внутриклеточных метаболических путей в зависимости от их непосредственных функциональных потребностей (Kumar & Dikshit 2019). В недавнем исследовании Sadiku с соавт. (2021) показали, что в условиях физиологического стресса (в отсутствие глюкозы и кислорода) для увеличения своей гликолитической способности и образования гликолитических промежуточных продуктов из неглюкозных субстратов нейтрофилы используют глюконеогенез. Авторы также показали, что, хотя для нейтрофилов характерен высокий исходный уровень окисления жирных кислот, при стимуляции медиаторами воспаления происходит значительное увеличение внутриклеточного гликолитического потока и окислительно-восстановительной буферной способности (Sadiku et al. 2021). Таким образом, дефекты во внутриклеточном цикле глюкозы могут негативно влиять на ключевые функции нейтрофилов, тогда как непропорциональное усиление гликолиза из-за чрезмерной доступности внеклеточной глюкозы поддерживает их постоянную активность (Sadiku et al. 2021).
Thimmappa с соавт. (2022) выявляли специфические реакции нейтрофилов на различные активирующие сигналы путем оценки дифференцированного фосфорилирования белков с использованием объективного количественного фосфопротеомного подхода. Авторы указали, что ex vivo воздействие гипергликемии на нейтрофилы в течение трех часов вызывало шестикратное усиление образования ВНЛ, в то время как при стимуляции либо липополисахаридом (ЛПС) (бактериальная инфекция), либо метаболическим промежуточным соединением гомоцистеином за тот же период оно было трехкратным. Избыточному образованию ВНЛ в условиях гипергликемии предшествовала конститутивная активация нейтрофилов (рис. 3B). Это наблюдение подтверждается различными исследованиями на животных, показывающими, что ожирение предрасполагает нейтрофилы к спонтанному выбросу слабых ВНЛ без стимуляции (Cichon et al. 2021, Burczyk et al. 2022). Считается, что в этом процессе участвует киназная активность ГТФазы Rho, индуцированная C-Jun-N-концевой киназой (JNK); известно, что эта активность играет важную роль в активации НАДФH-оксидазы, а также в регуляции перестройки актинового цитоскелета — оба этих события необходимы для образования ВНЛ (Lacy & Eitzen 2008, Gavillet et al. 2018, Thimmappa et al. 2022). Экстернализованные компоненты ВКЛ, которые включают хроматин, цитруллинированные гистоны, внеклеточную ДНК, нуклеосомы и NE, сохраняются в сосудистой сети и связаны с патогенезом прогрессирования заболевания (Kaplan & Radic 2012, Papayannopoulos 2018). В контексте диабета индуцированное гипергликемией конститутивное образование слабых ВНЛ считается основной патофизиологической причиной диабетической болезни почек (ДБП).
Исследования как на животных, так и с участием людей показали корреляцию между постоянным образованием ВНЛ и тяжестью ДБП (Gupta et al. 2022). В условиях высокого уровня глюкозы ВНЛ способствуют активации NLRP3 (NOD-like receptor protein 3)-инфламмасомы в фагоцитарных макрофагах, что, в свою очередь, усиливает воспаление за счет активности интерлейкина (IL)-1β, вызывая нарушение барьерной функции эндотелиальной/гломерулярной фильтрации и фиброз (Gupta et al. 2022). Сопутствующее повреждение сосудов микроциркуляторного русла вызывает выделение во внеклеточное пространство DAMP, которые включают нуклеиновые кислоты, аденозинтрифосфат (АТФ) и различные метаболиты из поврежденных эндотелиальных клеток, которые, в свою очередь, распознаются PRR на лейкоцитах, что приводит к гипервоспалительной реакции (Garcia-Martinez et al. 2015). Исследования на животных показали, что подавление образования ВНЛ с помощью ингибитора пептидиларгининдеиминазы 4 (PAD4) для предотвращения цитруллинирования гистонов и последующей дестабилизации хроматина в нейтрофилах может уменьшить дисфункцию эндотелия (Gupta et al. 2022) и что ингибирование активности NLRP3-инфламмасомы уменьшает тяжесть ДБП (Zhang et al. 2019a). Сходным образом, различные исследования также указывают на активность нейтрофилов при диабетической ретинопатии (He et al. 2022), остром повреждении легких (Jiang et al. 2014), периферической невропатии (Chen et al. 2021) и язвах стопы (Lee et al. 2020). В совокупности эти исследования подтверждают роль конститутивной активности нейтрофилов в прогрессировании заболеваний в условиях стерильного метаболического воспаления.
Однако в клинических условиях сам по себе контроль уровня глюкозы недостаточен для предотвращения вторичного повреждения тканей, вызванного персистирующим метаболическим воспалением. В рандомизированном контролируемом исследовании Menegazzo с соавт. (2018) показали, что терапия инсулином или дапаглифлозином (ингибитором натрий-глюкозного котранспортера 2 [SGLT2]) у пациентов с СД2 не способна подавить образование ВНЛ, несмотря на достижение контроля уровня глюкозы, тогда как применение метформина может снижать концентрации компонентов ВНЛ в плазме у пациентов с преддиабетом вне зависимости от контроля уровня глюкозы, путем предотвращения мембранной транслокации протеинкиназы C-βII и активации НАДФН-оксидазы. Также было показано, что противовоспалительные свойства метформина, проявляющиеся независимо от контроля уровня глюкозы, вызывают снижение ОНЛ на 9% у пациентов с СД2 после продолжительной (8–16 месяцев) терапии, тогда как этот феномен не наблюдался у пациентов, получавших монотерапию производными сульфонилмочевины, несмотря на достижение аналогичных уровней контроля уровня глюкозы (Cameron et al. 2016). В последующем исследовании Тихонова с соавт. (2020) показали, что гипергликемия у лиц с СД2 вызывает нарушения механизмов активации НАДФН-оксидазы и внутриклеточных Са2+-сигнальных систем в нейтрофилах. Таким образом, авторы подтвердили результаты Menegazzo с соавт. (2018) и предположили, что НАДФH-оксидаза в гранулоцитах может быть многообещающей мишенью для клинического вмешательства при лечении осложнений диабета, связанных с метаболическим воспалением. В настоящее время в фармацевтической промышленности разрабатывается несколько небольших молекул, направленных на ингибирование чрезмерной активности НАДФН-оксидазы, но, к сожалению, из-за нескольких ненаправленных эффектов и пагубных последствий полного блокирования продукции АФК очень немногие из этих молекул перешли в фазу I клинических испытаний (Altenhöfer et al. 2015, Sassetti et al. 2021).
Кроме того, неясно, восстановит ли блокирование постоянной активности НАДФН-оксидазы нормальные функциональные реакции нейтрофилов. Несмотря на высокий гликолитический потенциал этих лейкоцитов и их способность окислять жирные кислоты, непрерывная активация нейтрофилов в условиях высокого уровня глюкозы ограничивает реакцию на последующую стимуляцию ЛПС и нарушает хемотаксис (Joshi et al. 2020, Roy et al. 2022, Thimmappa et al. 2022). Задержка трафика нейтрофилов и неспособность обеспечить достаточный фагоцитарный ответ вместе с ограничением производства дополнительных, более сильных ВНЛ из-за этого «истощенного фенотипа» делают пациентов с диабетом уязвимыми для инфекции.
Моноциты/макрофаги
Моноциты/макрофаги играют критическую роль в разрешении воспаления, и нарушение их регуляции вовлечено в различные патологические состояния, связанные с диабетом, включая нарушение функции жировой ткани, атеросклероз, незаживающие раны и заболевания почек. Привлечение циркулирующих моноцитов и резидентных тканевых макрофагов к местам воспаления инициируется выделением хемокинов и внеклеточных везикул апоптотических клеток в составе ткани. Апоптотические везикулы (из апоптотических нейтрофилов и ткани) играют важную роль во внутриклеточной коммуникации и, как считается, содержат липиды, белки, микроРНК и иммуномодулирующие ферменты, которые вместе с DAMP влияют на статус активации макрофагов (Torr et al. 2012, Grant et al. 2019, Ross et al. 2021). Хорошо известно, что макрофаги обладают континуумом различных фенотипов, которые могут либо способствовать воспалению (M1 активируются классическим путем; регулируются индуцируемым гипоксией фактором 1 — HIF-1, NF-kB, сигнальным путем интерферон-регуляторного фактора) или — восстановлению/ремоделированию тканей (M2 активируются альтернативным путем; регулируются рецепторами, активируемыми пролифератором пероксисом — PPAR, ядерным фактором, связанным с эритроидом 2, сигнальным путем STAT6). Классически активируемые макрофаги (М1) (поверхностные маркеры CD14, CD80, CD86, CD38) для производства энергии в основном полагаются на гликолиз, тогда как альтернативно активируемые макрофаги (М2) (поверхностные маркеры CD36, CD206, CD163) имеют более высокий уровень окислительного фосфорилирования (Mantovani et al. 2004, Jha et al. 2015) (рис. 3A). Однако фенотипическая пластичность макрофагов очень сложна, и их способность переключать фенотип зависит не только от активирующих сигналов, но также от внутриклеточной биоэнергетики и доступности питательных веществ (Jha et al. 2015). Таким образом, эти клетки могут играть различные промежуточные роли в спектре между состояниями активации M1 и M2 в зависимости от факторов микроокружения.
При острой воспалительной реакции активность короткоживущих нейтрофилов сменяется активностью макрофагов М1. Классически активированные макрофаги являются фагоцитарными/цитотоксическими и имеют высокий уровень гликолиза (Jha et al. 2015, Rasheed & Rayner 2021). Они помогают устранять тканевый детрит (апоптотические тельца) и играют решающую роль в распознавании апоптотических нейтрофилов и удалении мертвых клеток в процессе эффероцитоза (Mahmoudi et al. 2022). Этот процесс рассматривается в других работах, но известно, что он включает (I) распознавание мертвых/гибнущих клеток с помощью сигналов find-me, которые включают растворимые хемокины, нуклеотиды и мембранные липиды; (II) фагоцитарные макрофаги реагируют на сигналы eat-me, такие как потеря асимметрии фосфолипидов на плазматической мембране, появление белков просвета эндоплазматического ретикулума на поверхности клетки и присутствие липидных биомолекул (лизофосфатидилхолин, LPC) на апоптотических тельцах и (III) поглощает/уничтожает апоптотические клетки для повторного использования клеточных компонентов и восстановления гомеостаза (Mahmoudi et al. 2022, Tajbakhsh et al. 2022). В ходе эффероцитоза происходит молекулярное переключение, при котором происходит фенотипическое и биоэнергетическое изменение макрофагов от М1 (высокая гликолитическая активность) к М2 (окислительное фосфорилирование) для устранения воспаления. Точный механизм такого переключения фенотипа до сих пор неясен, но считается, что он связан с активностью различных специализированных про-разрешающих липидных медиаторов (SPM) (таких как эйкозаноиды, липоксины, резолвины, протектины, марезины) (Ryan & Godson 2010, Börgeson & Godson 2012) и сигнальных путей с участием ядерных рецепторов (PPARγ/α, X-рецепторы печени, глюкокортикоидные рецепторы, орфанные ядерные рецепторы [Nur]) (Stifel et al. 2022). Во время этого процесса лизосомная деградация апоптотических телец посредством катаболизма погибших клеток обеспечивает субстраты для переключения обмена веществ в сторону окислительного фосфорилирования и запускает различные ядерные транскрипционные факторы, чтобы индуцировать транскрипцию иммуносупрессивных цитокинов, таких как IL-10 и трансформирующий фактор роста-β (Zhang et al. 2019b). Нарушение эффероцитоза, наблюдаемое при метаболических патологиях, таких как диабет, приводит к накоплению некротических/пироптотических телец нейтрофилов, которые усугубляют воспаление за счет повышенной активации макрофагов M1, дендритных клеток и лимфоцитарных реакций Th1/Th17 (Lee et al. 2022, Mahmoudi et al. 2022). Поскольку внутриклеточный метаболизм важен в определении функции макрофагов, помимо непрерывных сигналов активации, наблюдаемых при ожирении и диабете, из-за метаболических нарушений также происходят аберрантная активация и перепрограммирование макрофагов (An et al. 2019, Mahmoudi et al. 2022). Однако в настоящее время не хватает фармацевтических агентов, направленных на митохондриальный метаболизм макрофагов, чтобы способствовать разрешению воспаления. Важно отметить, что на биологию макрофагов влияют клеточное происхождение и локализация в ткани и что уникальные тканеспецифичные популяции макрофагов могут по-разному реагировать на нарушение регуляции метаболизма (Artyomov et al. 2016). Макрофагам жировой ткани (МЖТ) принадлежит центральная роль дисфункции жировой ткани; они чувствительны к длительным метаболическим изменениям.
В исследовании Kratz с соавт. (2014) с помощью протеомного подхода продемонстрировали, что макрофаги, полученные из жировой ткани при ожирении, не имеют ни фенотипа M1, ни M2, а скорее пребывают в состоянии метаболической активации. Авторы подтвердили свои результаты, поместив макрофаги в условия, характерные для метаболического синдрома (высокий уровень глюкозы, пальмитата, инсулина), и наблюдали метаболически активированное состояние, а не классические воспалительные фенотипы (Kratz et al. 2014). В подтверждение этого Boutens с соавт. (2018) исследовали метаболические признаки МЖТ у худых и тучных мышей и показали, что ожирение перепрограммирует макрофаги на «двойную» биоэнергетику для усиления как гликолиза, так и окислительного фосфорилирования. С помощью модели ex vivo, в которой макрофаги совместно культивировали с эксплантатами жировой ткани, авторы выяснили, что метаболическая активация макрофагов дозозависимо запускается факторами, образующимися в жировой ткани (Boutens et al. 2018). Постоянное воздействие избыточного лептина в условиях ожирения активирует JAK2–STAT3 и фосфатидилинозитол-3-киназу, а также сигнальные каскады мишени рапамицина у млекопитающих (PI3K–Akt–mTOR), что приводит к метаболическим изменениям, которые увеличивают поглощение глюкозы, усиливают регуляцию гликолитических ферментов, нарушают функцию митохондрий, а впоследствии — повышают фагоцитарную активность и выработку провоспалительных цитокинов (Monteiro et al. 2019). Boutens с соавт. (2018) в своем исследовании с помощью транскрипционного анализа подтвердили, что гликолиз был основным фактором выделения цитокинов в метаболически активированных макрофагах.
При болезненных состояниях с высокой метаболической потребностью предпочтительным источником для производства энергии служит обмен глюкозы, и, таким образом, гликолиз преобладает над окислительным фосфорилированием в явлении, известном как эффект Варбурга (Rasheed & Rayner 2021). Быстрое производство ацетил-КоА во время гликолиза способствует расширению эндоплазматического ретикулума и аппарата Гольджи для увеличения выработки провоспалительных цитокинов. Кроме того, считается, что усиленный гликолитический сигналинг, который опосредуется как доступностью субстрата, так и усилением транслокации переносчика глюкозы (GLUT)-1 на мембрану, формирует у макрофагов M1-подобный фенотип посредством эпигенетических изменений (Rasheed & Rayner 2021). Они связаны с взаимодействием гликолитических промежуточных продуктов, таких как пируваткиназа M2 и лактат, с гистондеацетилазами, вызывающими ремоделирование хроматина и, в конечном итоге, иммунную память, способствующую приобретению макрофагами провоспалительного фенотипа (Guzik & Cosentino 2018, Rasheed & Rayner 2021). Эпигенетические изменения метаболических путей в макрофагах пациентов с СД2, кроме того, влияют на динамику экспрессии TLR4 и способность этих клеток реагировать на последующие активирующие сигналы (Davis et al. 2020).
Эпигенетические изменения и иммунная память сохраняются даже после значительной потери веса и достижения контроля уровня глюкозы. В исследовании на животных Blaszczak с соавт. (2020) показали, что у мышей, получавших диету с высоким содержанием жиров в течение трех месяцев с последующим переходом на обычную диету в течение трех месяцев, память об ожирении сохранялась на тканевом уровне. Несмотря на значительную потерю массы тела, у этих животных не нормализовались иммунные клетки и сохранялось воспаление жировой ткани. Сходным образом, исследование, сравнивающее профиль SPM у больных диабетом с морбидным ожирением и пациентов с легким ожирением без диабета до и после бариатрической хирургии, показало, что, несмотря на значительную потерю веса и ремиссию диабета, у пациентов с диабетом сохранялось метаболическое воспаление (Schulte et al. 2020).
Кроме того, считается, что секретом нерегулируемых МЖТ влияет на взаимодействие между адипоцитами и опухолевыми клетками. В исследовании ex vivo Vallega с соавт. (2022) изучали влияние макрофагов на ранние стадии развития опухоли при ожирении. Для исследования паракринных взаимодействий авторы использовали человеческую систему совместного культивирования и продемонстрировали, что среда, кондиционированная провоспалительными макрофагами, усиливает взаимодействие между клетками рака молочной железы и адипоцитами, в результате чего клетки рака молочной железы становятся более агрессивными (Vallega et al. 2022). Точные молекулярные механизмы, лежащие в основе этого взаимодействия, до сих пор неясны, но, вероятно, они многофакторны и связаны с обменом дисфункциональными метаболитами, непропорциональной продукцией цитокинов и факторов роста и переносом внеклеточного везикулярного карго (Vallega et al. 2022, Li et al. 2023). Необходимо лучшее понимание свойств и функций МЖТ с нарушениями регуляции при ожирении–диабете. До тех пор, пока новые методы лечения не станут доступными, раннее вмешательство на предиабетической стадии, направленное на контроль воспаления (например, противовоспалительные средства) и вторичного клеточного повреждения (например, антиоксиданты), имеет решающее значение для предотвращения начала эпигенетических изменений и долговременной иммунной памяти в макрофагах.
Дендритные клетки, тучные клетки, базофилы, инвариантные натуральные киллеры
Считается, что некоторые другие, менее известные клетки врожденного иммунитета, такие как дендритные клетки, тучные клетки и базофилы, играют патологическую роль в развитии ожирения и резистентности к инсулину (Wu & Van Kaer 2013, Park et al. 2018, Żelechowska et al. 2018, Zatterale et al. 2019). Исследования на животных ясно продемонстрировали накопление провоспалительных клеток врожденного иммунитета, в том числе тучных клеток, дендритных клеток и базофилов, в жировой ткани во время ожирения, вызванного диетой с высоким содержанием жиров (Liu et al. 2009, Cho et al. 2016, McLaughlin et al. 2017). Базофилы и тучные клетки в первую очередь отвечают за защиту хозяина от паразитарных инфекций и играют роль в аллергических реакциях, тогда как дендритные клетки являются специализированными антигенпрезентирующими клетками, которые связывают врожденную и адаптивную иммунные системы, представляя антигены лимфоцитам (Gasteiger et al. 2017).
Накопление тучных клеток и дендритных клеток в ВЖТ связано с провоспалительным статусом жировой ткани и косвенно влияет на привлечение и активацию нейтрофилов, макрофагов и лимфоцитов посредством выделения цитокинов/хемокинов (Żelechowska et al. 2018). Влияние гипергликемии на тучные клетки человека было продемонстрировано в исследовании in vitro, которое показало, что высокий уровень глюкозы способствует фосфорилированию киназы, регулируемой внеклеточными сигналами — JNK, и митоген-активируемой протеинкиназы p38 для увеличения внутриклеточной продукции воспалительных цитокинов, таких как TNFα, IL-1β и IL-6 (Nagai et al. 2012). Накопление тучных и дендритных клеток в соединительной ткани, ассоциированной с жировой, также влияет на здоровый рост жировой ткани (дифференцировку преадипоцитов), изменяет выделение ключевых липидных медиаторов, усиливает экспрессию белков внеклеточного матрикса/фиброз и способствует общей дисфункции жировой ткани и последующей резистентности к инсулину (Żelechowska et al. 2018, Aldan et al. 2019, Zatterale et al. 2019). Это было подтверждено на животных моделях, показывающих, что нокаут дендритных или тучных клеток снижает инфильтрацию макрофагов в жировую ткань и ослабляет резистентность к инсулину в условиях ожирения (Liu et al. 2009, Cho et al. 2016). Напротив, регуляторные клетки, такие как ILC2s и iNKTs, чувствительны к липидной перегрузке в жировой ткани, и при активации их ответ направлен на уменьшение воспаления (Park et al. 2018). Однако у людей с ожирением эти клетки истощены и поэтому не могут выполнять свою иммуномодулирующую функцию. В совокупности эти данные подтверждают идею о том, что иммунная активация и активация воспалительных цитокинов начинаются во время преддиабетического состояния ожирения; однако необходимы дальнейшие исследования, чтобы понять, как длительное воздействие патологического микроокружения, связанного с СД2, влияет на функцию этих менее заметных клеток врожденного иммунитета.
Заключение
Следует расширить клиническое ведение пациентов с диабетом так, чтобы не только сосредоточиться на снижении веса и контроле уровня глюкозы, но и включить в него независимую противовоспалительную стратегию для улучшения общего исхода и замедления прогрессирования заболевания. Хотя обсуждение потенциальных терапевтических стратегий и эффективности различных фармацевтических молекул выходит за рамки данного обзора, он подчеркивает необходимость лечения этого состояния, лежащего в основе заболевания у пациентов с СД2. Из-за сложностей и нарушения метаболической и иммунной регуляции с участием множества факторов, связанных с вялотекущим метаболическим воспалением, доклинические исследования только сейчас начинают проливать свет на потенциальные терапевтические мишени. Таким образом, в наших знаниях существует множество пробелов, связанных с неспособностью разрешить воспаление и долговременной врожденной иммунной памятью. На основании современных данных рекомендуется раннее вмешательство на преддиабетической стадии ожирения для предотвращения аберрантной активации лейкоцитов, эпигенетических изменений и долговременной иммунной памяти.
