
Клеточная биоэнергетика: экспериментальные данные об адаптации, вызванной алкоголем

Аннотация
Чрезмерное употребление алкоголя связано с мультисистемными эффектами и поражением органов-мишеней; также оно вносит значительный вклад в глобальное бремя болезней. Было выявлено несколько механизмов с участием алкоголя; при этом биоэнергетическая дезадаптация привлекает как основной патофизиологический механизм, способствующий повреждению клеток. В этом обзоре, основанном на фактических данных, основное внимание уделяется современным знаниям о вызванных алкоголем биоэнергетических адаптациях в метаболически активных тканях и органах: печени, сердечной и скелетной мышцах, поджелудочной железе и головном мозге. Метаболизм алкоголя сам по себе существенно влияет на биоэнергетические пути в тканях, особенно в печени. Алкоголь подавляет дыхательные состояния в цепи переноса электронов, а также активность и экспрессию дыхательных комплексов, в результате чего снижается содержание АТФ. Кроме того, алкоголь нарушает регуляцию основных метаболических путей, включая гликолиз, цикл трикарбоновых кислот и окисление жирных кислот. На эти биоэнергетические нарушения влияют опосредованные алкоголем изменения в морфологии, биогенезе и динамике митохондрий. В обзоре подчеркиваются сходства и различия в биоэнергетической адаптации в зависимости от типа ткани, модели (острой или хронической) употребления алкоголя и доступности энергетического субстрата. Нарушенная биоэнергетика действует синергично с другими критическими патофизиологическими механизмами, включая повышенный окислительный стресс, и ускоряет клеточную дисфункцию, способствуя старению, регулируемой гибели клеток и повреждению органов-мишеней.
Введение
Чрезмерное употребление алкоголя является самой дорогостоящей формой употребления психоактивных веществ, при этом глобальные экономические издержки оцениваются примерно в 1300 долларов на одного взрослого, из которых 38% приходится на прямые затраты, а 60% связаны с потерей производительности [1]. Чрезмерное употребление алкоголя значительно снижает ожидаемую продолжительность жизни, связано с потерей более чем двух миллионов лет потенциальной жизни и является фактором, связываемым с 5,3% всех смертей ежегодно [2]. Патофизиология потребления алкоголя сложна, начиная от риска травм и от острой интоксикации до кумулятивного повреждения органов и тканей в результате хронического рискованного употребления алкоголя, а также значительного влияния на психическое здоровье лиц с расстройствами, связанными с употреблением алкоголя (РСУА) [3–5]. Однако тяжесть вызванного алкоголем повреждения тканей и прогноз различаются у разных людей и зависят от таких факторов, как генетика, обмен веществ, возраст, пол, этническая принадлежность, окружающая среда и образ жизни [4].
Чрезмерное употребление алкоголя определяется как употребление более трех или четырех порций алкоголя в определенный день или более 7 или 14 порций алкоголя в неделю для женщин и мужчин, соответственно [6]. Запойное пьянство — модель употребления алкоголя, при которой концентрация алкоголя в крови повышается до 0,08% или выше (80 мг/дл), как правило, в результате употребления 4–5 алкогольных порций в течение двух часов, также относится к категории высокого риска [6]. Доля взрослого населения, злоупотребляющего алкоголем, значительна: примерно 18% взрослых во всем мире сообщают об эпизодическом употреблении алкоголя в больших количествах в течение последнего месяца, а 6,6% взрослого населения Соединенных Штатов сообщают о чрезмерном употреблении алкоголя [7].
B индуцированное алкоголем множественное повреждение органов вовлечено несколько механизмов, включая метаболизм алкоголя, окислительный стресс, дисбиоз кишечника и иммунную активацию, воспаление, некроз и регулируемую гибель клеток, перестройку внеклеточного матрикса и эпигеномную адаптацию [8–22]. Недавние данные подтверждают важность биоэнергетической адаптации как основного патофизиологического механизма, общего для тканей, поражаемых алкоголем, который достигает высшей точки в создании метаболической нестабильности и повреждении органов-мишеней. Этот основанный на фактических данных обзор посвящен исключительно современным знаниям о вызванных алкоголем биоэнергетических адаптациях в метаболически активных тканях. Обзоры механизмов алкогольного повреждения тканей см. в [23–30]. Обзоры митохондриальной адаптации, опосредованной алкоголем, см. в [31–38].
Роль биоэнергетических изменений при повреждении тканей, вызванном алкоголем
Биоэнергетика в строгом смысле описывается как реакции окисления, происходящие вместе с прохождением электронов через белковые комплексы и коферменты митохондриальной мембраны и сопряженные с синтезом АТФ. Такая хемиосмотическая гипотеза окислительного фосфорилирования (OXPHOS) была предложена Питером Митчеллом в 1961 году [39]. С тех пор число биоэнергетических и метаболических исследований экспоненциально росло по мере развития современных методов проведения биоэнергетических измерений и их интеграции с данными метаболомных, протеомных и эпигеномных исследований. Этот подход значительно улучшил понимание того, как биоэнергетика механически модулирует состояние здоровья и развитие заболеваний. В основе клеточной биоэнергетики лежит митохондрия — «энергетическая станция клетки».
Адаптация клеток млекопитающих к энергетическим ресурсам и потребностям позволила развиться митохондриям с митохондриальной ДНК (мтДНК), сохранившей основные гены, контролирующие производство энергии. В зависимости от клеточных потребностей каждая клетка имеет от нескольких сотен до тысяч копий мтДНК [40]. Потребность клетки в энергии удовлетворяется за счет сложной связи между митохондриями, ядром и цитоплазмой на основе доступности субстрата, восстанавливающих эквивалентов и выработке активных форм кислорода (АФК) [41].
Клеточный метаболизм энергетических субстратов (глюкоза, жиры и аминокислоты) генерирует АТФ, необходимый для удовлетворения потребности в энергии, и регулирует окислительно-восстановительное состояние клеток. Глюкоза окисляется до пирувата и в клетках, содержащих митохондрии, образует ацетил-КоА. Жирные кислоты, с другой стороны, преобразуются в ацетил-КоА через митохондриальный путь β-окисления. Глюкогенные аминокислоты могут быть преобразованы в глюкозу, пируват или промежуточное соединение цикла трикарбоновых кислот (ЦТК), а кетогенные аминокислоты способны превращаться в жир, ацетил-КоА или ацетоацетил-КоА. Ацетил-КоА входит в цикл трикарбоновых кислот с образованием никотинамидадениндинуклеотида + водорода (НАДН). НАДН поступает в электрон-транспортную цепь (ЭТЦ) и окисляется комплексом I (КI) путем последовательного переноса электронов (e−) на флавинмононуклеотид, железо-серные кластеры (Fe–S) и кофермент Q. Сукцинат — еще один промежуточный продукт ЦТК — окисляется КII, перенося e− на флавинадениндинуклеотид (ФАД), Fe–S и кофермент Q. Цитохром-с-редуктаза (КIII), с другой стороны, принимает e− в два этапа, где убихинон (CoQ) и убихинол (CoQH2) связываются с КIII, и этот цикл повторяется еще раз (Q-цикл). Наконец, e− переносится на кислород с помощью КIV (цитохром-с-оксидазы) [42]. Когда e− проходит через окислительно-восстановительные протонные насосы (КI, III и IV), вместе с высвобождением энергии также происходит транспорт протонов (H+) через внутреннюю мембрану митохондрий для создания на ней электрохимического потенциала. Когда уровень АТФ низкий, Н+ проникает в матрикс через АТФ-синтазу с образованием АТФ, который затем экспортируется в цитоплазму транслокаторами адениновых нуклеотидов (АNТ) [43]. Поскольку такой процесс фосфорилирования АДФ, приводящий к образованию АТФ, требует кислорода, он известен как OXPHOS (рисунок 1). Процесс митохондриального синтеза АТФ критически зависит от поглощения Ca2+ митохондриальным комплексом захвата кальция [44–48]. Кроме того, элегантные исследования в лаборатории Sollott показали, что АТФ-синтаза также служит митохондриальным переносчиком K+. Авторы предположили, что за синтез АТФ отвечает транспорт как K+, так и Н+ [49, 50]. Эффективность, с которой ЭТЦ производит АТФ, называется эффективностью сопряжения. В системе ЭТЦ с высоким уровнем сопряжения на каждую израсходованную калорию вырабатывается максимальное количество АТФ. Наоборот, в менее сопряженной системе для производства того же количества АТФ требуется более высокое потребление калорий [41].
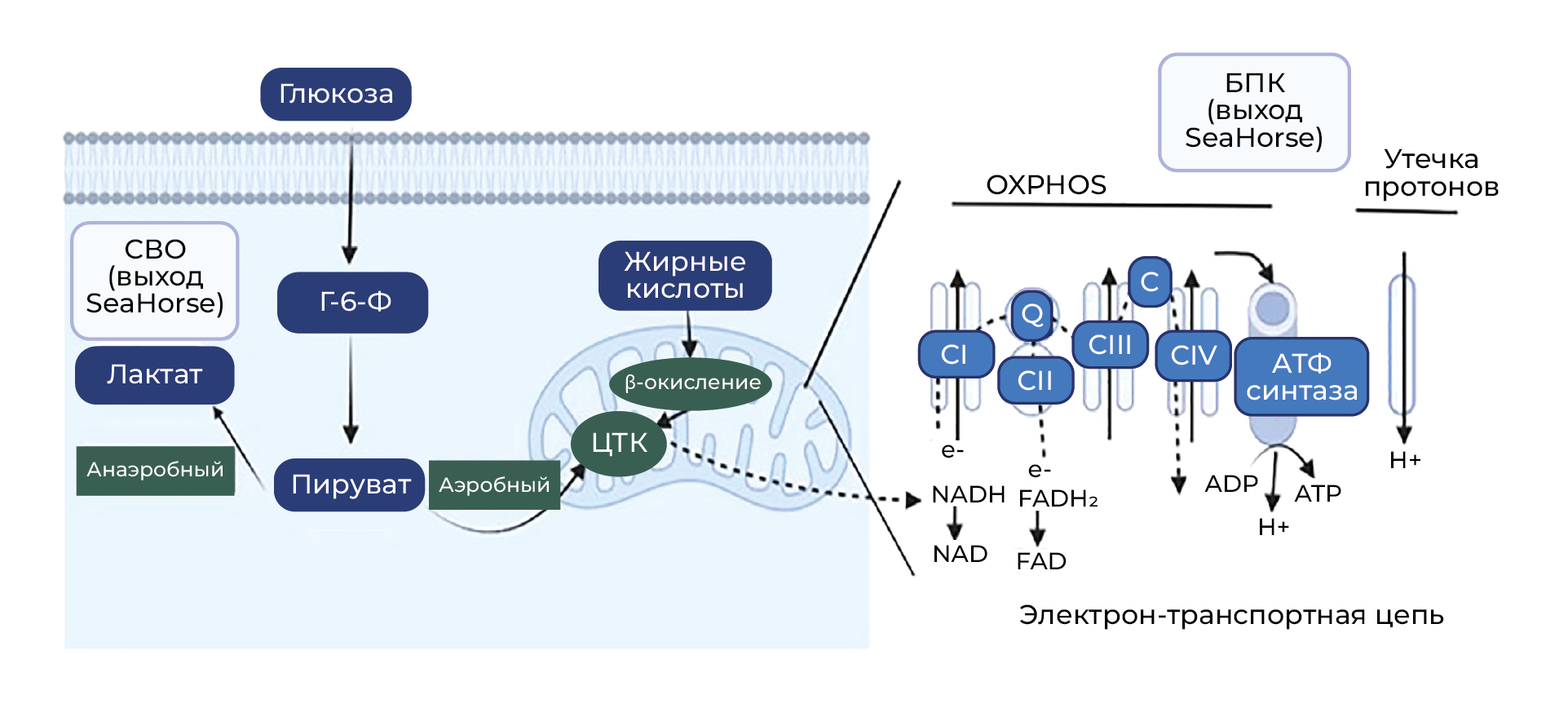
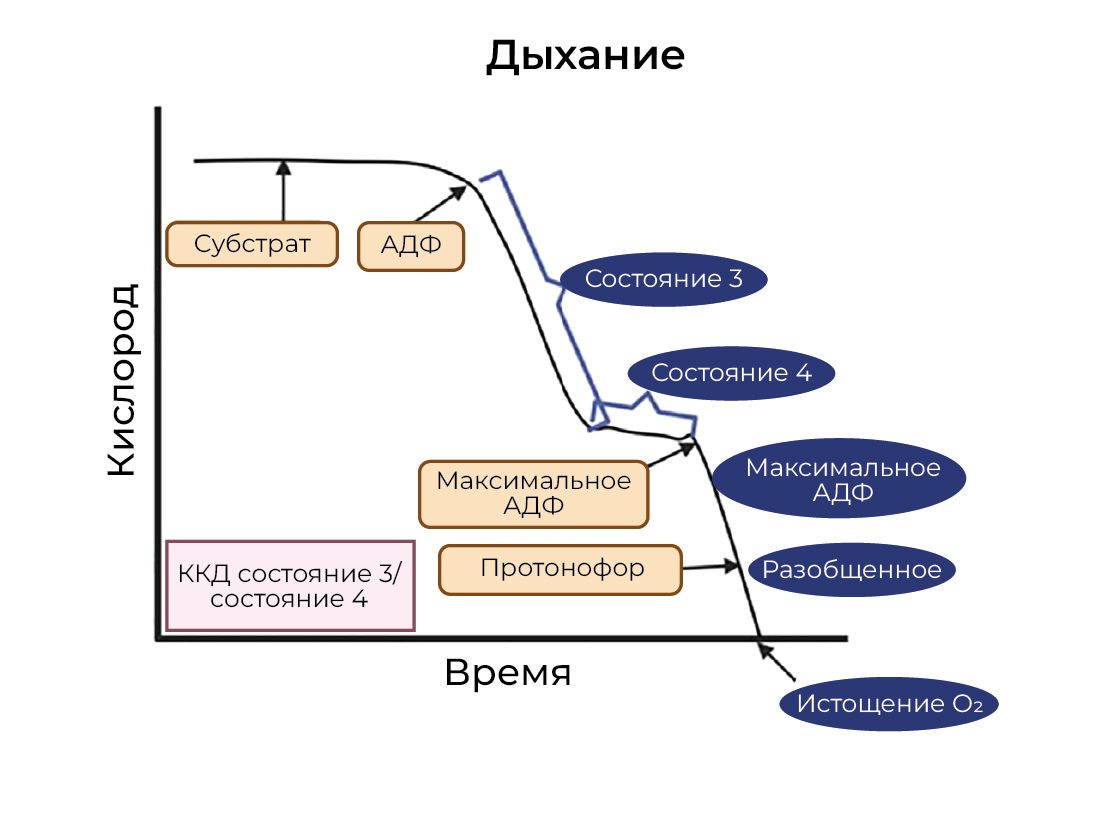
Chance и Williams описали пять состояний митохондриального дыхания, используя протокол оксиграфии (рисунок 2).
- Состояние 1 достигается, когда уровни АДФ и субстрата низкие и частота дыхания низкая.
- Состояние 2 достигается, когда добавляется высокая концентрация АДФ, но скорость дыхания низкая, поскольку уровни субстрата низкие.
- Состояние 3 индуцируется при добавлении субстратов, и вместе с уже имеющейся высокой концентрацией АДФ наблюдается низкое соотношение АТФ/АДФ и высокое потребление кислорода (JO2) [51].
- Состояние 4 характеризуется высоким соотношением АТФ/АДФ и низким уровнем АДФ. Уровень JO2 низкий, так как потребление кислорода происходит исключительно за счет утечки протонов [52].
- Состояние 5 указывает на пероксильные радикалы (ROX) или аноксию [53]. Увеличение частоты дыхания в состоянии 4 или снижение коэффициента дыхательного сопряжения (т. е. 3 JO2 / состояние 4 JO2) позволяет предположить снижение эффективности сопряжения [54].
Оксислительно-восстановительное (редокс-) состояние представляет собой баланс между АФК, образующимися во время клеточного метаболизма, и антиоксидантной системой, которая нейтрализует АФК. Митохондриальные АФК содержат неспаренный e−, и при частичном восстановлении молекулярного кислорода образуются супероксид (O2•−) и перекись водорода (H2O2) [55, 56]. O2•− и H2O2 могут реагировать с ионами переходных металлов, способствуя дальнейшему образованию радикалов, включая гидроксильный радикал (•OH) [41, 57]. Более того, снижение электрохимического потенциала на внутренней митохондриальной мембране индуцирует пору митохондриальной проницаемости (mtPTP) — механизм саморазрушения, который активируется повышением уровня Ca2+ в митохондриальном матриксе или АФК [41]. Существует несколько антиоксидантных механизмов для подавления избыточного образования АФК. Митохондриальный НАДФH + H+ с помощью глутатионпероксидаз восстанавливает окисленный глутатион (GSSG) до глутатиона (2GSH) [41]. Сходным образом, окисленный тиоредоксин-S2 восстанавливается НАДФН и тиоредоксинредуктазой [58]. Матричная супероксиддисмутаза, содержащая Mn (MnSOD), и Cu/ZnSOD (также присутствует во внутренней митохондриальной мембране), превращают O2•− в H2O2. Кроме того, митохондриальный тиоредоксин-2 взаимодействует с митохондриальным пероксиредоксином-3, снижая уровень АФК [59]. Таким образом, по мере того, как окислительно-антиоксидантный баланс смещается от антиоксидантной защиты клетки к продукции АФК, повреждение липидов, белков и нуклеиновых кислот приводит к окислительному стрессу, повреждению клеток и, в конечном итоге, к клеточной гибели [34].
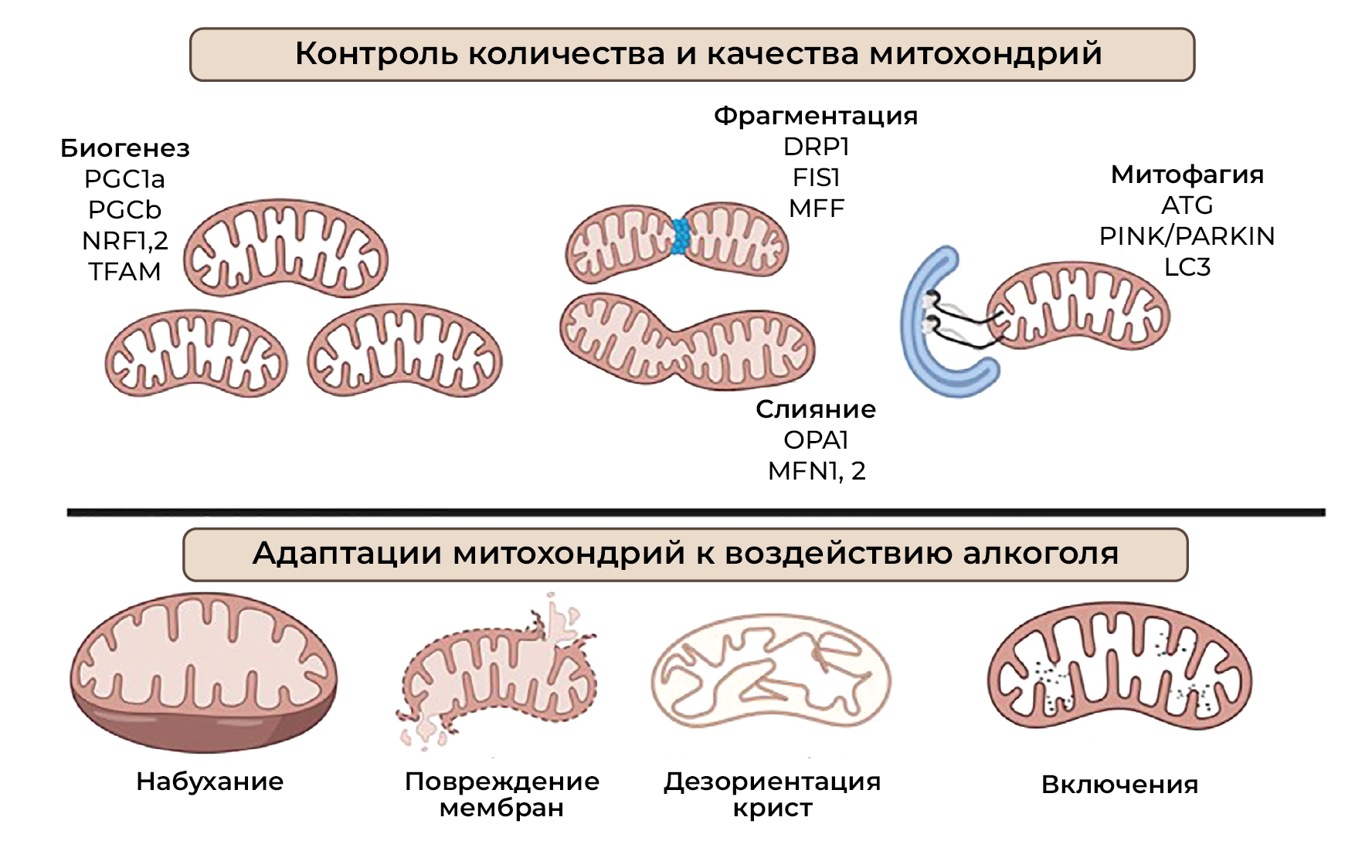
Общая биоэнергетическая способность клетки зависит от числа и качества митохондрий (рисунок 3). Биогенез митохондрий позволяет увеличить их размер и количество и регулируется γ-коактиватором 1-/α и b (PGC-1α/b) рецептора, активируемого пролифератором пероксисом (PPAR), котранскрипционными факторами и его взаимодействиями с транскрипционными факторами/белками, такими как как ядерные респираторные факторы (NRF-1 и NRF-2) и митохондриальный транскрипционный фактор А (TFAM). Биогенез митохондрий в совокупности модулируют разобщающие белки (UCP2), PPAR, тиреоидный гормон, глюкокортикоид, эстроген и связанные с эстрогеном рецепторы α и γ [60, 61]. Исследования показывают, что биоэнергетика также неразрывно связана с динамикой митохондрий, включая события слияния и деления/фрагментации [62–64]. В слиянии митохондрий участвуют белок 1 атрофии зрительного нерва (OPA1) и митофузины 1 и 2 (MFN1 и MFN2) [65–67]. Деление/фрагметация митохондрий регулируется белком, родственным ГТФазе динамину 1 (DRP1) [68], фактором фрагментации митохондрий (MFF) [69] и белком фрагментации митохондрий 1 (FIS1) [70]. Кроме того, MFN2 играет важную роль в OXPHOS [71–73], а снижение уровня MFN1 и OPA1 влияет на потенциал митохондриальной мембраны, дыхание и уменьшает стабильность суперкомплексов ЭТЦ [74–76]. Как обсуждалось, митохондрии чувствительны к АФК, и процессы восстановления митохондрий, включая митофагию, имеют решающее значение для предотвращения распространения окислительного стресса. Опосредованные алкоголем воздействия на митохондриальный биогенез, динамику и митофагию рассмотрены в других работах [30, 31, 34, 77–81].
Экспериментальная оценка клеточной биоэнергетики
Чтобы лучше интерпретировать вызванные алкоголем изменения в клеточной биоэнергетике, важно иметь общее представление о доступных в настоящее время инструментах. Анализ путей обмена веществ обеспечивает высочайшее разрешение для биоэнергетических измерений за счет использования распределения меченых изотопомеров для измерения внутренних потоков метаболитов [82]. Кроме того, биоэнергетические анализы в реальном времени с применением фосфоресцентных зондов, измеряющих внешние потоки, и в сочетании с протоколами метаболических субстратов и ингибиторов позволяют проводить биоэнергетические измерения в короткие временные промежутки. Эти популярные анализы с низким молекулярным разрешением включают респирометрию и анализ внеклеточного потока (EFA) [83]. Респирометрия — потенциометрический метод с применением платиновых и серебряных электродов (Clark Oxygraph-2 K; Oroboros) — используется наиболее часто [84, 85]. Революционная технология Seahorse (Agilent Technologies) измеряет потребление кислорода и выделение протонов в очень небольшом объеме среды непосредственно над клеточным монослоем (переходная микрокамера) с помощью сенсорных зондов (www.seahorsebio.com). Это также позволяет последовательно добавлять определенные метаболические ингибиторы для динамического измерения клеточной биоэнергетики и функции митохондрий [40, 61, 86, 87]. Недостаток большинства из этих систем заключается в том, что они используют экстремальные условия анализа и могут не воспроизвести баланс свободной энергии, доступной in vivo [88]. В настоящем обзоре кратко обсуждаются результаты, полученные с использованием этих тестов.
Респирометрию с помощью электродов Кларка проводят на пермеабилизированных клетках или изолированных митохондриях. Для измерения дыхательных состояний отслеживают потребление кислорода в присутствии митохондриальных субстратов (глутамат/малат — субстраты комплекса I; сукцинат — субстрат комплекса II) с АДФ или без него (рисунок 2) [89]. OroborosO2k (Oroboros Instruments) — двухкамерный респирометр с полярографическими датчиками кислорода, позволяющий калибровать кислород и интегрировать результаты с помощью программного обеспечения DatLab. Это позволяет проводить респирометрию с высоким разрешением; технология развивалась с годами, позволяя устанавливать новые стандарты в биоэнергетических исследованиях. Недостаток по сравнению с XF96 заключается в том, что параллельно можно анализировать только два образца, но можно проводить функциональное исследование отдельных комплексов митохондриальной дыхательной цепи [90, 91].
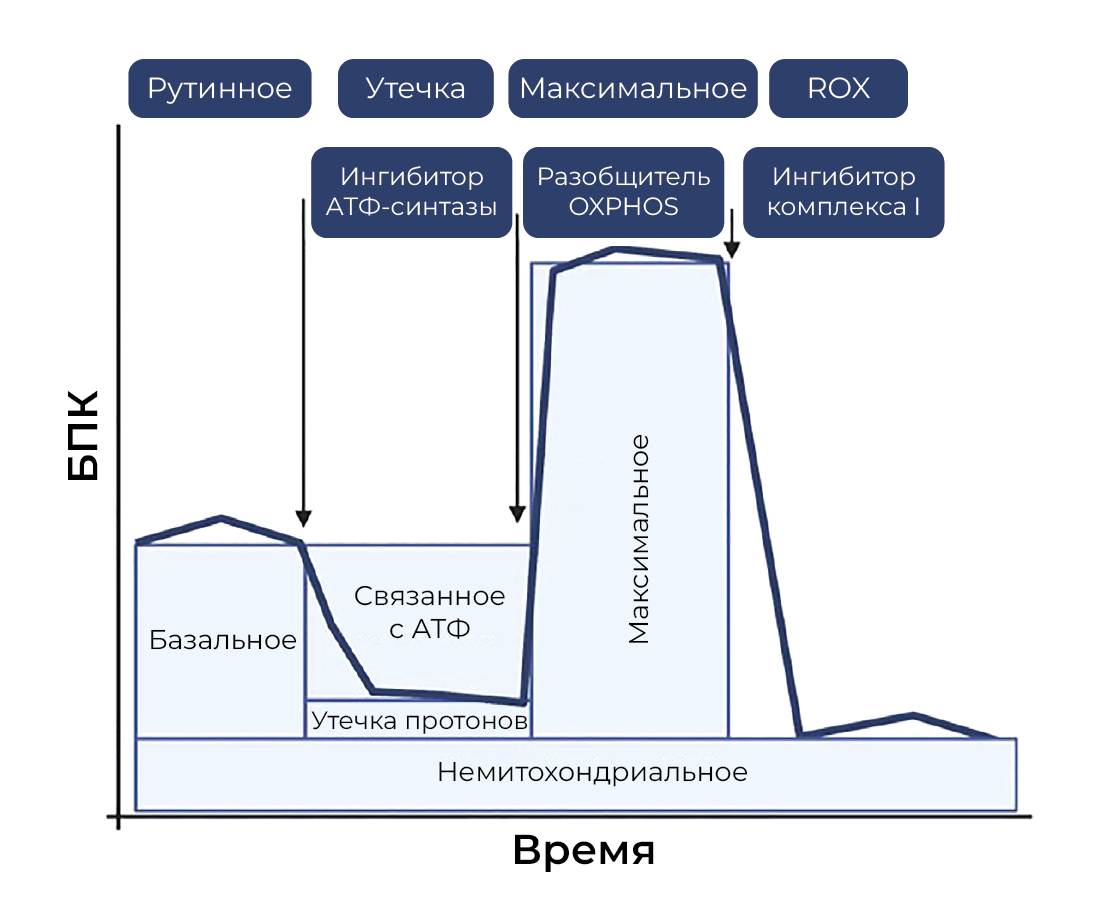
XF96 EFA — для измерения потребления кислорода с помощью популярного стресс-теста Mito можно использовать интактные клетки или изолированные митохондрии. Этот анализ измеряет (A) базальную скорость потребления кислорода (БПК): способность клетки удовлетворять базовые потребности в энергии и указывает на потребность в АТФ из-за утечки протонов; (Б) производство АТФ: измеряется путем введения олигомицина, ингибитора АТФ-синтазы, и отражает основное дыхание, которое обеспечивает производство АТФ; (В) утечку протонов: базальное дыхание, не связанное с производством АТФ. Повышенная утечка протонов может указывать на менее эффективно работающие митохондрии. (Г) Максимальная БПК измеряется путем добавления карбонилцианида 4-(трифторметокси)фенилгидразона — мощного разобщителя митохондриального OXPHOS. Снижение БПК отражает дефицит биогенеза митохондрий, повреждение мтДНК или аппарата дыхания или ограничения доступности или транспорта субстрата. Другие измерения включают (Д) резервную емкость: отражает способность клетки реагировать на потребности в энергии и (Е) немитохондриальное БПК: измеряет немитохондриальное потребление кислорода после добавления ротенона (ингибитор КI) и антимицина (ингибитор КIII). Как правило, это указывает на образование АФК или другие процессы потребления кислорода, включая активность провоспалительных ферментов. Также можно измерить активность комплекса дыхательной цепи [40] и индекс биоэнергетического здоровья (ИБЗ) — log [(резервная емкость × АТФ-связанное БПК)/немитохондриальное БПК × БПК утечки протонов] [92] (рисунок 4). Хотя технология SeaHorse обеспечивает высокую пропускную способность скрининга, удобна для пользователя и позволяет проводить измерения в небольших количествах образцов, измерения Oroboros более чувствительны и точны. Тем не менее, в обе эти технологии вносятся значительные улучшения, чтобы имитировать физиологические условия и сделать их надежными системами для биоэнергетических измерений [93, 94].
Скорость внеклеточного окисления (СВО), измеренная либо потенциометрически (EFA), либо с помощью флуоресцентных органических катионов, является мерой образования лактата, отражающего уровень гликолиза. Другие измерения включают оценку окислительно-восстановительного потенциала НАДН с помощью автофлуоресценции [97, 98]; определение активности ферментов [97]; определение градиентов pH и ионов с использованием флуорометрических зондов [99,100]; генетически кодируемые зонды для измерения изменений внутриклеточного pH, соотношения GSH/GSSG и окислительно-восстановительного состояния НАД(Ф)H [88].
Алкоголь и его метаболиты способствуют биоэнергетической адаптации
Знаковые исследования метаболизма алкоголя проводились в начале 1900-х годов. Нобелевскую премию по медицине 1955 года получил доктор Хьюго Теорелл за ценный вклад в изучение метаболизма алкоголя, включая разработку ферментативного метода его определения. Алкоголь метаболизируется практически во всех тканях, причем наибольший процент окисления алкоголя приходится на печень, что создает большую метаболическую нагрузку на этот орган [22]. Хотя энергетическая ценность алкоголя составляет 7 ккал/г, в отличие от других энергетических субстратов, он не запасается в тканях и остается в содержащейся в организме воде до элиминации. В отличие от других макронутриентов, скорость элиминации алкоголя не регулируется гормонально [22].
Алкоголь метаболизируется в цитоплазме с помощью алкогольдегидрогеназы (АДГ) до ацетальдегида с образованием НАДН. Реокисление комплекса АДГ-НАДН является лимитирующей стадией окисления этанола, поскольку образующийся НАДН должен поступать в митохондрии для окисления в ЭТЦ [101]. Поскольку NADH не способен проходить через мембраны, для проникновения в митохондрии он использует малатно-аспартатный или α-глицерофосфатный челнок. Таким образом, окислительно-восстановительное состояние клеток во время метаболизма алкоголя зависит как от челночной активности, так и от эффективности ЭТЦ. В митохондриях ацетальдегид превращается в ацетат под действием ацетальдегиддегидрогеназы (АлДГ), также образуя НАДН и способствуя снижению отношения НАД+/НАДH. Высокие концентрации ацетальдегида, 0,6–1,5 мМ, (зарегистрированные уровни в крови находятся в диапазоне мкМ [102, 103]) являются мощными ингибиторами челноков, а внемитохондриальная система окисления НАДН менее чувствительна к ацетальдегиду. Кроме того, ацетальдегид ингибирует транспорт глутамата, фосфата и цитрата, которые служат составными частями челноков, снижая их эффективность для транспорта НАДН [104]. Таким образом, общее ингибирование НАДН-челноков, вызванное ацетальдегидом, позволяет предположить, что выработка ацетальдегида может быть лимитирующим фактором метаболизма этанола.
Производство ацетата в результате окисления этанола, которое катализируется АДГ1, происходит в основном в печени; на него приходится большая часть потребления кислорода печенью [105]. Ацетат может поступать в метаболические пути, подобные тем, в которые вступают углеводы, жиры и белки. Однако высокий уровень НАДH, образующегося при окислении этанола в печени, препятствуют вхождению ацетата в ЦТК из-за восстановления цитоплазматического и внутримитохондриального НАД [106]. НАД+ представляет собой кофермент, необходимый во многих метаболических путях, включая гликолиз, ЦТК, OXPHOS; также он требуется для работы пируватдегидрогеназного комплекса. Ферменты гидридного переноса катализируют восстановление НАД+ до НАДН. Затем митохондриальный НАДН используется в ЭТЦ и служит субстратом для производства АТФ. Таким образом, общее снижение отношения НАД+/НАДН ослабляет клеточный окислительный метаболизм [107]. Соотношение НАД+/НАДН также регулирует окислительно-восстановительное состояние внутри клетки, а увеличение количества НАДН, образующегося при окислении алкоголя, приводит к повышению уровня АФК. Снижение окислительно-восстановительного отношения NAD+ /NADH и образование ацетальдегида снижает уровень митохондриального глутатиона (mGSH) и уменьшает антиоксидантный резерв клетки. Это вызванное алкоголем снижение уровня mGSH частично связано с нарушением транспорта GSH через внутреннюю митохондриальную мембрану [108]. Кроме того, хотя хроническое воздействие этанола не изменяет окислительно-восстановительное отношение в цитоплазме, в митохондриях оно снижается [109]. Этанол снижает уровень пирувата в печени [110] и, как следствие, уменьшение активности пируваткарбоксилазы ослабляет глюконеогенез [111]. Все эти метаболические изменения в конечном итоге усиливают синтез жирных кислот в печени, что вместе с повышенным поглощением липидов приводит к их накоплению в печени [111]. Ацетат, образующийся в результате метаболизма этанола, может использоваться в качестве энергетического субстрата тканями — в основном мозгом, скелетными и сердечными мышцами. Повышенные концентрации ацетата приводят к снижению β-окисления длинноцепочечных жирных кислот — предпочтительного энергетического субстрата в сердечной и скелетной мышцах [112, 113]. Кроме того, высокий уровень ацетата также усиливает ацетилирование белков и влияет на экспрессию генов за счет усиления ацетилирования гистонов, что, в свою очередь, может изменить метаболические и другие ключевые функциональные пути в определенных тканях [111]. Таким образом, общее снижение соотношения НАД+/НАДH и истощение пула антиоксидантов ингибируют ключевые метаболические пути, включая гликолиз, ЦТК, окисление жирных кислот (ОЖК) и глюконеогенез, что приводит к снижению продукции АТФ (рисунок 5).
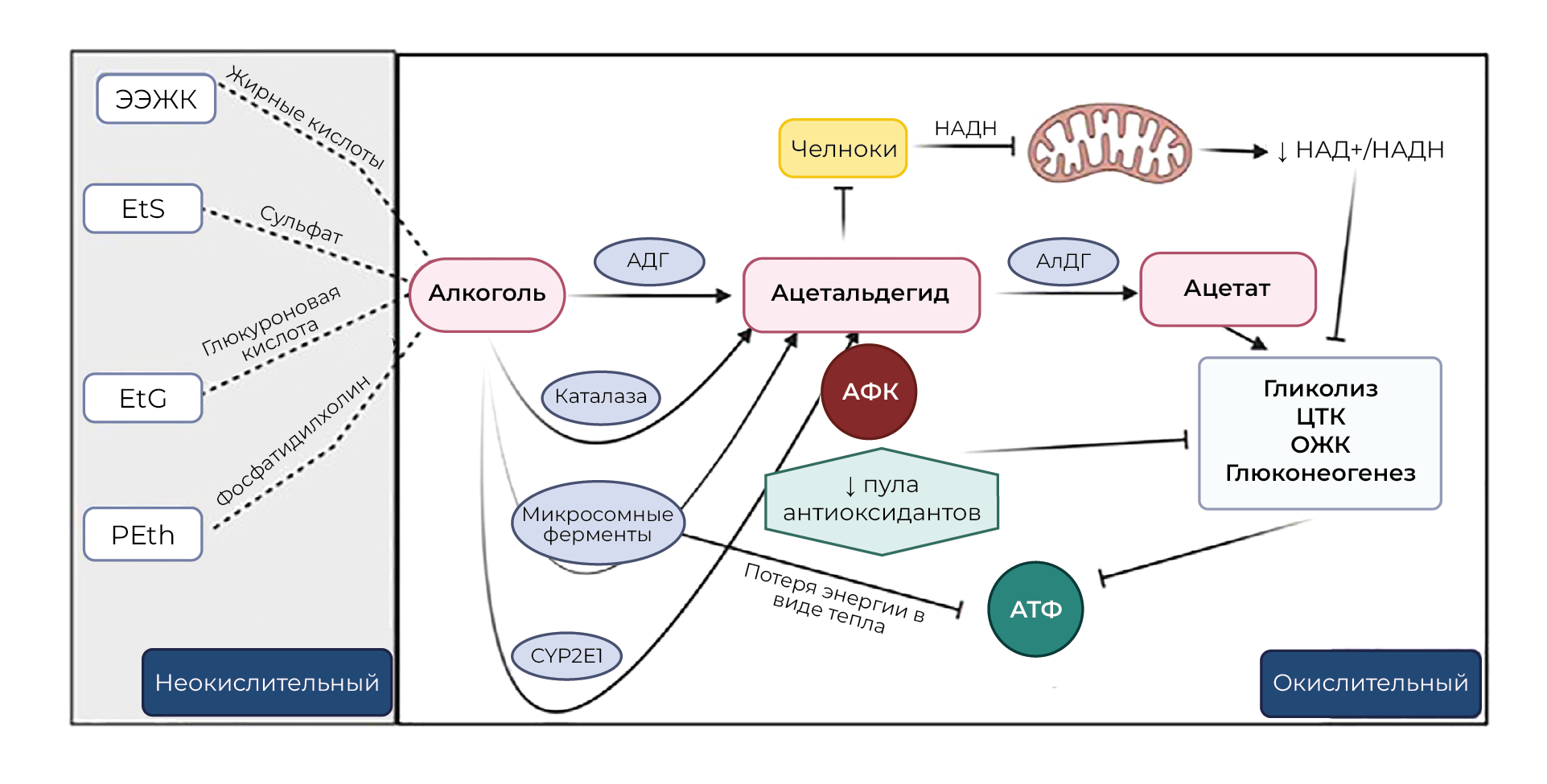
Хроническое злоупотребление алкоголем индуцирует дополнительные пути его метаболизма, включая печеночный цитохром p450 2E1 (CYP2E1). Метаболизм алкоголя через эту систему приводит к образованию значительного количества АФК и нарушению антиоксидантных механизмов [114–116]. Кроме того, в печени и других тканях активируются микросомальные ферменты [117, 118], что приводит к неэффективному сопряжению окисления и фосфорилирования и потере чистой энергии в результате теплопродукции [119]. Отчасти метаболизм алкоголя происходит под действием каталазы в пероксисомах, хотя это второстепенный окислительный путь. Небольшая часть алкоголя может метаболизироваться неокислительными путями, что приводит к ферментативной конъюгации с эндогенными метаболитами (жирными кислотами, фосфолипидами, сульфатами и глюкуроновой кислотой), что приводит к образованию этиловых эфиров жирных кислот (ЭЭЖК), фосфатидилэтанола, этилсульфата (EtS) и этилглюкуронида (EtG). Образование ЭЭЖК катализируется синтазой ЭЭЖК, присутствие которой особенно характерно для головного мозга, сердца, печени и поджелудочной железы [120] (рисунок 5).
Таким образом, метаболизм алкоголя, особенно в печени, приводит к биоэнергетическим изменениям, которые потенциально могут способствовать вызываемому алкоголем повреждению клеток, как описано в следующем разделе. Окислительный стресс, опосредованный метаболизмом алкоголя, также представляет собой основной механизм вызываемого алкоголем повреждения тканей, что рассмотрено в других работах [30, 34, 36, 77, 121, 122].
Алкоголь модулирует клеточный метаболизм
Опубликованные данные о влиянии алкоголя на энергетический обмен датируются серединой ХХ века. В основополагающих исследованиях Ammon и Estler с использованием острого и хронического введения алкоголя мышам основной причиной функционального повреждения клеток считалось снижение содержания АТФ, а не прямое токсическое действие алкоголя [123]. Позже это было подтверждено в доклинических моделях с использованием хронического употребления этанола [124–127]. Значение метаболитов алкоголя было подтверждено исследованиями, показавшими, что ацетальдегид в очень высоких концентрациях снижает синтез АТФ в митохондриях печени [128, 129].
Таким образом, понимание вызываемых алкоголем изменений клеточного метаболизма основано на данных доклинических моделей в сочетании с воздействием алкоголя на клетки и ткани ex vivo. Здесь кратко описаны наиболее часто используемые модели хронического употребления алкоголя и их характерные особенности. Самостоятельное введение жидкой алкогольной диеты, чаще всего алкогольной диеты Либера-ДеКарли, обеспечивающей 100% суточной калорийности, сравнивают с контрольной диетой такой же калорийности [130]. К другим моделям относят парадигму «питья в темноте», когда животные имеют ограниченные периоды доступа к алкоголю в воде в темное время суток в течение нескольких дней или недель [131]. Системное введение алкоголя проводится, как правило, с помощью внутрибрюшинных инъекций [132, 133] или внутрижелудочных (ВЖ) инфузий через хирургически имплантированные ВЖ зонды (например, модель Цукамото–Френча [134]) или через желудочный зонд, чтобы избежать влияния вкуса. Кроме того, для изучения воздействия алкоголя на повреждение тканей были выведены генетически модифицированные мыши или крысы с большей или меньшей чувствительностью к алкоголю или предпочтением к нему [135, 136]. Ни один из этих методов не дает идеального отражения характера, количества или частоты употребления алкоголя человеком. Тем не менее, каждая модель дает представление о различных патофизиологических изменениях тканей и органов, возникающих в результате различных моделей воздействия алкоголя. Более того, объединение результатов этих исследований in vivo с результатами, полученными при обработке клеток или тканей этанолом ex vivo и in vitro, позволяет исследовать механизмы этих процессов и создавать гипотезы.
Биоэнергетическая адаптация, опосредованная алкоголем: данные по метаболически активным тканям
Печень
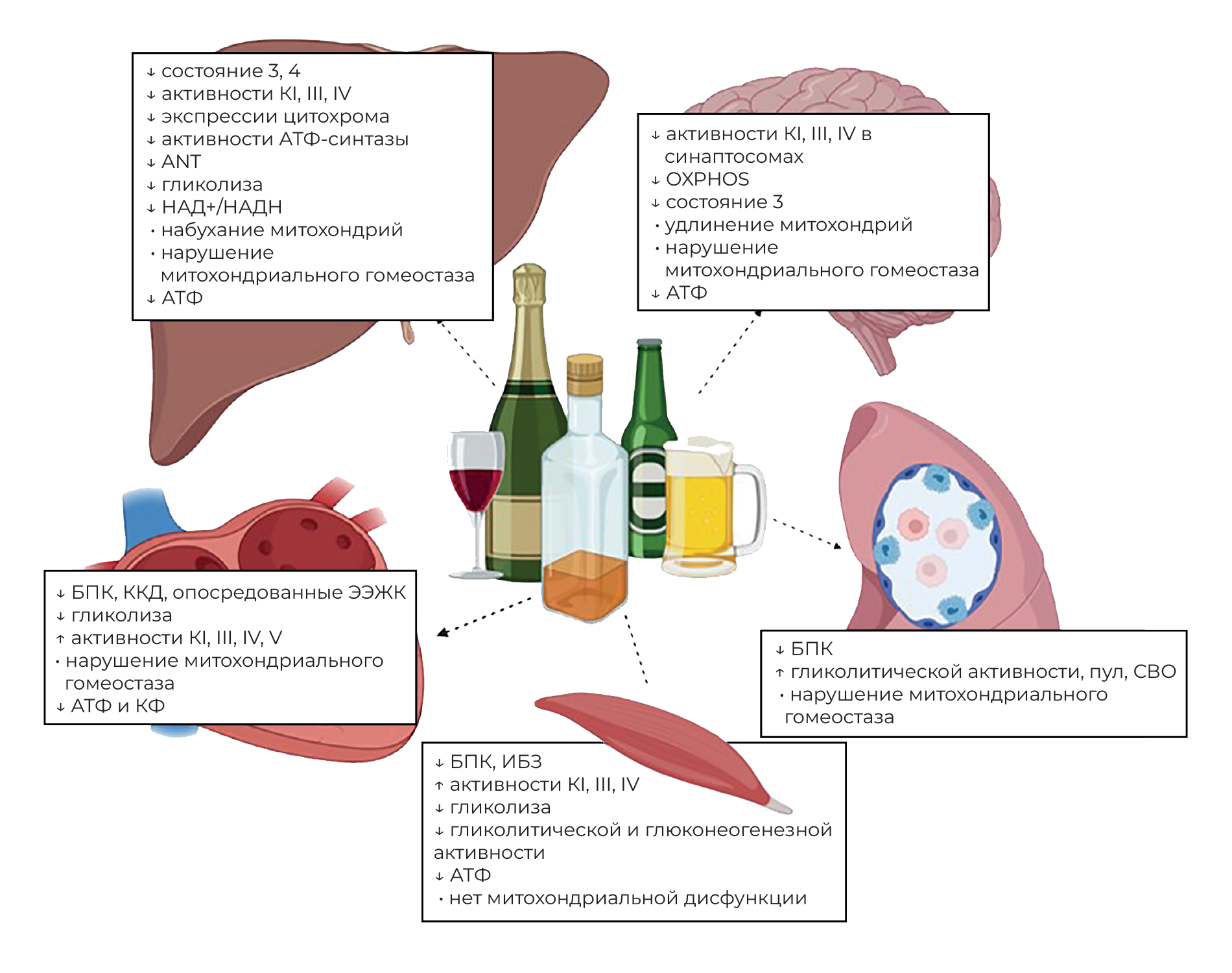
Клетки паренхимы печени богаты митохондриями и являются основными местами метаболизма алкоголя. Результаты ex vivo, in vitro, доклинических и клинических исследований продемонстрировали, что изменения стадий дыхания в ЭТЦ, дыхательных комплексов, гликолиза, содержания АТФ, митохондриальной морфологии и мутаций мтДНК инициируют алкогольную болезнь печени и способствуют ее прогрессированию (рисунок 6).
Воздействие алкоголя, особенно хроническое, снижает уровень биоэнергетического дыхания в гепатоцитах и выделенных митохондриях печени. Хроническое употребление алкоголя снижает уровень как сопряженного, так и несопряженного дыхания за счет прямого воздействия на само дыхание и эффективность сопряжения с сопутствующим снижением скорости синтеза АТФ [137, 138]. Выделенные митохондрии печени крыс, употреблявших алкоголь с питьевой водой в течение десяти недель или получавших алкогольную диету, имели значительно сниженный уровень дыхания в состоянии 4 без значительных изменений в протонной движущей силе и OXPHOS в сайте сопряжения II. Эти исследования показали, что алкоголь влияет на снижение активности компонентов ЭТЦ, которые контролируют сопряженное дыхание и не влияют на разобщенное, — например, цитохром-с-оксидазы [139, 140]. В выделенных митохондриях печени крыс, которые получали спиртовую диету Либера-ДеКарли, снижался уровень дыхания в состоянии 3, когда использовались несколько субстратов, и это было связано со снижением активности КIII и АТФ-синтазы [141, 142]. Thayer и Rottenberg также показали, что алкоголь снижает частоту дыхания в состояниях 3 и 4 и коэффициент контроля дыхания (ККД) в митохондриях печени [143]. Сходным образом, митохондрии печени имели значительное снижение дыхания в состоянии 3 и ККД с повышением чувствительности к NO-зависимому ингибированию дыхания [144]. У павианов, получавших алкоголь, у которых развилась жировая дистрофия печени или продвинутые стадии фиброза, дыхание в состоянии 3 было снижено с уменьшением отношения АДФ/О и ККД для всех субстратов. Одновременно снижалась экспрессия цитохрома и активность глутамата, НАДН и сукцинатдегидрогеназы (СДГ) [145]. Даже умеренное употребление алкоголя (10% этанола в течение четырех недель) приводило к снижению дыхания в состояниях 3 и 4 с использованием в качестве субстратов как глутамат-малата, так и сукцината, снижению активности цитохром-с-оксидазы и содержания цитохрома аа3, повышению активности СДГ и увеличению Vmax активности ANT в митохондриях печени; это позволяет предположить, что любое количество потребляемого алкоголя увеличивает биоэнергетическую нагрузку [146]. Биоэнергетическая нагрузка связана с гипоксией, опосредованной алкоголем, и считается важным фактором, который способствует гепатотоксичности и некрозу как перивенозных, так и перипортальных гепатоцитов. Кроме того, гепатоциты крыс, получавших алкоголь, имели пониженную биоэнергетическую резервную способность митохондрий и большую чувствительность к NO-зависимому торможению дыхания как в нормальных, так и в гипоксических условиях, что подтверждает концепцию о том, что оксид азота и алкоголь повышают восприимчивость к гипоксии и усугубляют вызванное алкоголем повреждение печени [147].
Помимо прямого влияния алкоголя на дыхание, изменение экспрессии и активности ферментов также в значительной степени способствует подавлению дыхания. Хроническое воздействие алкоголя снижает экспрессию печеночных цитохромов а и b и активность цитохром-с-оксидазы [148, 149]. В печени самцов крыс, получавших алкогольную диету Либера-ДеКарли, была понижена экспрессия митохондриальных дыхательных комплексов I, III, IV и V. Это также было связано со снижением экспрессии ключевых генов, регулирующих функцию митохондрий и мтДНК, включая PGC-1α, NRF1, TFAM [150]. In vitro обработка этанолом гепатоцитов в течение 24 ч или выделенных митохондрий в течение 3 ч снижала ферментативную активность КI и КIV. Этанол также снижал активность транслоказы АДФ и экспрессию СДГ в гепатоцитах [151]. Как известно, митохондриальные ЭТЦ организованы в надмолекулярные комплексы, называемые респиросомами [152], которые не только увеличивают эффективность переноса между комплексами, но и уменьшают утечку электронов. Тем не менее, клетки, экспрессирующие митохондриально-направленный CYP2E1, имеют значительно более низкий уровень респиросом, и их истощение усугубляется алкоголем как in vivo, так и in vitro. Хотя большая часть CYP2E1 находится в эндоплазматическом ретикулуме (ЭПР), он в значительной степени экспрессируется в других органеллах, включая митохондрии [153, 154]. Содержание CYP2E1 в митохондриях увеличивается у животных, получавших алкоголь [155], и вместе с цитохром-с-оксидазой он играет критическую роль в опосредованном алкоголем митохондриальном дыхании. Хотя алкоголь значительно снижает уровни мтДНК и кодируемой митохондриальным геномом мРНК, возможно, это вторичные эффекты. Более того, почти полное восстановление респиросом и устранение повреждений ДНК митохондриально-направленными антиоксидантами и ингибиторами CYP2E1 предполагает их потенциал в качестве терапевтических мишеней для борьбы с токсичностью алкоголя и повреждением тканей [156]. Объединение данных, полученных от мышей с нокаутом активирующего транскрипционного фактора 4 (ATF4), пациентов с алкогольным гепатитом и специфичной гиперэкспрессией TFAM в печени, показала, что активация ATF4 служит ключевым медиатором стресса ЭПР, который нарушает биогенез митохондрий и дыхательную функцию с участием TFAM-опосредованных путей [38].
Путь введения алкоголя, по-видимому, оказывает незначительное дифференциальное влияние на биоэнергетическую адаптацию митохондрий печени. Например, у крыс, которые самостоятельно получали жидкую алкогольную диету, наблюдалось снижение дыхания в состоянии 3 с глутаматом/малатом, ацетальдегидом и сукцинатом в качестве субстратов, ККД и экспрессии КI, III, IV и V. Однако внутрижелудочная (ВЖ) инфузия алкоголя увеличивала дыхание с глутаматом/малатом и ацетальдегидом в качестве субстратов, соотношение НАД+/НАДН и уменьшало дыхание с сукцинатом в качестве субстрата [35]. Эти отличия могут быть связаны с различной концентрацией алкоголя, которая достигается при ВЖ введении и при употреблении алкоголя в жидкой диете. ВЖ введение также сопровождалось учащением дыхания с ацетальдегидом в качестве субстрата, несмотря на снижение уровня белков комплекса I и III. Это может быть связано с явлением, известным как митохондриальный пороговый эффект (избыток и резерв мРНК, тРНК и комплексов дыхательной цепи) [157]. Оно служит защитным механизмом, особенно при наличии мутаций мтДНК и в присутствии хронических стрессоров, таких как алкоголь. Кроме того, различные реакции наблюдаются в зависимости от видов животных, используемых при экспериментальном введении алкоголя. У крыс, по-видимому, происходит избыточная экспрессия дыхательных белков, что может объяснить несоответствие в корреляции между уровнями белков дыхательных комплексов и митохондриальным дыханием. При этом у мышей экспрессия дыхательных белков не усиливается, и существует сильная корреляция между митохондриальным дыханием и экспрессией дыхательных белков [35]. Как самостоятельное пероральное употребление алкоголя, так и ВЖ вливание усиливали митохондриальное дыхание при использовании глицерол-3-фосфата (который доставляет электроны от цитоплазматического НАДH в митохондрии) и октаноата (субстрата для β-окисления) [35]. Аналогичным образом, биоэнергетическую адаптацию также может модулировать состав рациона. У крыс, получавших диету с высоким содержанием жиров и достаточным количеством белка, хроническое воздействие алкоголя снижало уровень дыхания в состояниях 3 и 4 как с малатно-глутаматным, так и с сукцинатным субстратом. Интересно, что эти изменения не наблюдались при диетах с высоким содержанием белка и низким содержанием жиров. При использовании НАД-связанных субстратов с высоким содержанием жира и достаточным количеством белка также значительно понижался ККД; при высоком содержании белка и низком содержании жира такой эффект не наблюдался [158]. Наконец, доступность субстрата определяет метаболическую функцию. В условиях ограниченного оборота пирувата алкоголь снижает уровень глюкозы и увеличивает выработку лактата. Однако в присутствии аланина (предшественника глюконеогенеза) алкоголь увеличивает общую выработку глюкозы у сытых животных, но снижает ее у животных натощак с сопутствующим увеличением содержания молочной кислоты [159].
Алкоголь также опосредует гликолитическую адаптацию, которая вместе с нарушением OXPHOS приводит к снижению выработки АТФ и последующей клеточной дисфункции. Однократное пероральное введение 5 г алкоголя на 1 кг массы тела снижало образование АТФ путем гликолиза и максимально усиливало поглощение кислорода в перфузируемой изолированной печени через 2,5 ч после введения (быстрое усиление метаболизма алкоголя) [160]. Такому снижению частично способствует само окисление спирта, особенно при истощении промежуточных соединений малатно-аспартатного челнока. Восстанавливающие эквиваленты, образующиеся при окислении этанола, конкурируют за перенос в митохондрии с эквивалентами, образующимися в ходе гликолиза и, таким образом, ингибируют аэробный гликолиз. Одновременно происходит уменьшение отношения НАД+/НАДН в цитоплазме, что ослабляет анаэробный гликолиз [161]р. Постоянное воздействие алкоголя нарушает гликолитический путь как в нормоксической, так и в гипоксической среде на стадии между глюкозой и глицеральдегид-3-фосфатом [162,163]. И, как обсуждалось для OXPHOS, в перивенозной области печени снижение напряжения кислорода из-за окисления этанола в значительной степени способствует уменьшению гликолитической выработки АТФ, что усиливает риск повреждения печени [164].
Являются ли морфологические изменения митохондрий результатом биоэнергетических адаптаций или прямого действия алкоголя, до конца не выяснено (рисунок 3). Митохондрии часто увеличены, имеют причудливую форму, дезориентированные кристы и паракристаллические включения. Однако содержание митохондриального белка на мтДНК остается неизменным, а морфологические изменения, по-видимому, отражают повреждение митохондриальной мембраны, а не адаптивную гипертрофию [145]. Предполагается, что алкоголь вызывает набухание митохондрий за счет увеличения проницаемости митохондриальной мембраны в два этапа. На первом этапе спирт мгновенно вызывает разбавление всей среды, а нарушение митохондриальной проницаемости (с помощью открытия пор — прим. перев.) позволяет перераспределить воду и спирт в зависимости от концентрации. На втором этапе происходит зависящее от времени набухание митохондрий при высоких концентрациях этанола. Значительный вклад в набухание митохондрий вносит нарушение градиента K+ [168]. Возможно, что биоэнергетическая адаптация под действием алкоголя опосредуются ремоделированием митохондрий. Оно включает изменения динамики деления/фрагментации и слияния; митофагию и биогенез, а также кальциевый сигналинг, продукцию АФК и эпигеномные изменения [169, 170]. Фактически, Han et al, используя модели внутрижелудочного и перорального введения алкоголя, предположили, что ремоделирование митохондрий имеет решающее значение для адаптаций митохондрий печени, управляющих биоэнергетическими изменениями в ответ на алкоголь [35]. Тем не менее, хотя в краткосрочной перспективе ремоделирование митохондрий может быть компенсаторным механизмом, хроническая нагрузка на печень в дальнейшем может привести к повреждению печени.
Хотя метаболизм алкоголя создает большую метаболическую нагрузку на печень, опосредованная алкоголем биоэнергетическая адаптация также наблюдается в метаболически активных тканях, включая сердечную и скелетную мускулатуру, поджелудочную железу и головной мозг (рисунок 6).
Сердечная и скелетная мускулатура
Сердечные и скелетные мышцы имеют высокие метаболические потребности и богаты митохондриями. В сердечной ткани митохондрии составляют ~ 35% объема [171] и в состоянии покоя покрывают ~ 90% потребности в АТФ. В скелетных мышцах митохондрии составляют около 3–8% объема [172], и на эту величину сильно влияет физическая активность. Таким образом, прямое или косвенное воздействие опосредованного алкоголем нарушения клеточной биоэнергетики приводит к дисфункции сердечной и скелетной мышечной массы.
Хроническое введение алкоголя собакам снижало содержание сердечной внутримитохондриальной изоцитратдегидрогеназы, АТФ, потребление кислорода митохондриями и ККД, что приводило к изменению работы миокарда [173, 174]. Сходным образом, у крыс, хронически получавших алкоголь, в сердечной мышце повышалась активность глюкозо-6-фосфатдегидрогеназы, альдолазы и глицеральдегидфосфатдегидрогеназы, но снижалась активность изоцитратдегидрогеназы [175]. В сердце ЭЭЖК представляют собой обычные метаболиты алкоголя, а митохондрии кардиомиоцитов, инкубированные с ЭЭЖК, снижают ККД и максимальные БПК. Кроме того, ЭЭЖК связываются с митохондриями; важно отметить, при инкубации с этанолом это происходит с 72% внутриклеточно синтезированных этиловых эфиров. Расщепление ЭЭЖК и образование жирных кислот приводит к разобщению OXPHOS; таким образом, ЭЭЖК могут действовать как токсический челнок для жирных кислот [176]. Острое воздействие алкоголя также влияет на биоэнергетику клеток сердца. Внутрибрюшинное введение алкоголя приводит к значительному увеличению гликолиза и ослаблению митохондриального дыхания и ККД в течение 30 мин; эти эффекты исчезают при понижении уровня алкоголя в крови и миокарде. Тем не менее хроническое внутрибрюшинное введение того же количества алкоголя подавляло гликолиз, уменьшало содержание гликогена, АТФ, креатинфосфата, митохондриальное дыхание и ККД изолированных митохондрий сердца [177]. Таким образом, оказывается, что острое воздействие алкоголя может вызывать компенсаторное усиление гликолиза для удовлетворения потребности в энергии. Напротив, хроническое воздействие алкоголя снижает как гликолитическое, так и митохондриальное дыхание, что может привести к значительному снижению продукции АТФ и, таким образом, к нарушению сердечной функции. Хроническое алкогольное воздействие также увеличивает экспрессию PPARα в сердце и снижает PGC-1α, что указывает на нарушение метаболизма жирных кислот в сердце. Также было выявлено снижение количества мРНК, белка и активности GAPDH, что говорит о нарушении выработки гликолитической энергии. Кроме того, увеличивалось содержание фруктозы, что указывает на компенсаторную адаптацию для активации альтернативных путей метаболизма глюкозы, таких как сорбитовый путь, для удовлетворения энергетических потребностей [178]. В отличие от печени, в сердечной ткани у животных, получавших алкоголь, наблюдается значительное увеличение активности митохондриальных ферментов (цитратсинтазы, КI, III, IV, V) и адаптивное увеличение количества мтДНК, свидетельствующее об увеличении числа митохондрий. Эти результаты предполагают тканеспецифические биоэнергетические адаптации в ответ на алкоголь [179].
Функционально митохондрии скелетных мышц имеют высокий ККД, соотношение АДФ/О и высокую скорость дыхания в состоянии 3 с различными субстратами. Тем не менее, неблагоприятное воздействие хронического злоупотребления алкоголем на митохондриальную функцию скелетных мышц неясно. С помощью объективного микрочипового анализа мышечных трубок, обработанных 100 мМ этанолом, было установлено, что дефекты компонентов ЭТЦ, эндогенных антиоксидантов и ферментов, участвующих в ЦТК, регулируются по-разному. Этанол in vitro также нарушал клеточное дыхание, снижал функцию комплексов I, II и IV и подавлял OXPHOS. Такое снижение содержания АТФ и окислительно-восстановительного отношения нарушало регуляцию окисления сукцината в ЦТК [180]. Однако Cardellach с соавт. [181] утверждают, что ассоциированная с употреблением алкоголя миопатия не связана со снижением энергоснабжения митохондрий. Мембранные препараты из печени и скелетных мышц, полученные из одних и тех же животных, потреблявших алкоголь, показали, что мембраны печени развили толерантность к воздействию алкоголя, в то время как мышечные мембраны сохранили к нему нормальную чувствительность. Авторы заключают, что отсутствие развития толерантности мышечных мембран коррелирует с отсутствием химических изменений в фосфолипидах, нормальной функцией митохондрий и саркоплазматического ретикулума, а также отсутствием дефицита митохондриальной энергии в скелетных мышцах [181]. С помощью биоптатов скелетных мышц людей с чрезмерным употреблением алкоголя было показано, что скорость окисления различными субстратами, активность комплексов дыхательной цепи и содержание цитохромов не изменились [182]. Аналогичным образом, Trounce и его коллеги сообщили о нормальном уровне мышечного гликогена, карнитина и нормальной активности митохондриальных ферментов-маркеров у людей, хронически злоупотребляющих алкоголем [183]. Однако они объяснили снижение активности гликогенолитических и гликолитических ферментов наблюдаемой атрофией волокон типа 2, которая обычно встречается при хроническом злоупотреблении алкоголем [182, 183]. Вопреки этому аргументу, в мышцах людей, злоупотреблявших алкоголем, активность цитохром-с-оксидазы и объемы митохондрий были ниже при более высоком содержании креатинфосфата [184]. Кроме того, обмен митохондриальным содержимым в результате событий деления/фрагментации и слияния уменьшалось как из-за генетических нарушений in vivo, так и вследствие хронического злоупотребления алкоголем. Был выявлен митофузин-1-зависимый путь, при котором ингибирование слияния снижало митохондриальный метаболический резерв и нарушало регуляцию колебаний концентраций кальция при длительной стимуляции. Таким образом, длительное подавление слияния ставит под угрозу биоэнергетику и сопряжение возбуждения и сокращения, обеспечивая потенциальный механизм, способствующий развитию алкогольной миопатии [185]. Следовательно, хотя есть предположения, что чрезмерное употребление алкоголя может не влиять на энергообеспечение митохондрий, опосредованное алкоголем нарушение биоэнергетики может в значительной степени способствовать непереносимости физической нагрузки, нарушению метаболического гомеостаза и регенеративной способности мышечных стволовых клеток.
Работа научной группы авторов обзора продемонстрировала, что хроническое злоупотребление алкоголем (ХЗA) индуцирует нарушение регуляции митохондриальных генов в скелетных мышцах на конечной стадии заболевания вирусом иммунодефицита обезьян (ВИО) у макак-резусов, не получавших антиретровирусную терапию (АРТ). ХЗА и AРT снижали активность СДГ в волокнах типа 1 и типа 2b и экспрессию гена PGC-1β в скелетных мышцах макак ХЗА/ВИО по сравнению с неинфицированным контролем. Более того, ХЗA предотвращало опосредованную ВИО-инфекцией активацию экспрессии генов, связанных с митофагией. Эти данные говорят о том, что инфекция ВИО нарушает митохондриальный гомеостаз и в сочетании с ХЗA приводит к дифференциальной экспрессии генов, участвующих в таком нарушении [186]. Более того, в миобластах, выделенных у макак ХЗA/ВИО/AРT+, было показано снижение максимального БПК, а формотерол, агонист β-адренорецепторов, частично его восстановил [187]. Результаты проведенных авторами доклинических исследований в настоящее время перенесены на изучение людей с дисгликемией, живущих с ВИЧ (ЛЖВ) и злоупотребляющих алкоголем. Полученные данные показывают, что более высокий балл в Тесте для определения расстройств, связанных с употреблением алкоголя (AUDIT), был связан с отрицательными показателями биоэнергетического здоровья, включая утечку протонов, немитохондриальное потребление кислорода и ИБЗ в скелетных мышцах ЛЖВ. Это также было связано с увеличением объема митохондрий и снижением экспрессии генов, участвующих в митохондриальной функции [188]. Помимо того, что биоэнергетические изменения могут регулировать метаболическую способность скелетных мышц, согласно более ранним данным авторов обзора, алкоголь-опосредованные сдвиги в биоэнергетическом фенотипе лежат в основе нарушения дифференцировки мышечных стволовых клеток. In vitro обработка этанолом первичных миобластов макак увеличивала максимальное БПК миобластов и снижала гликолитический метаболизм (СВО) дифференцировки D0, что было связано с опосредованным этанолом снижением индекса слияния, числа миотубок на поле и индекса дифференцировки миобластов. Кроме того, алкоголь нарушает гликолитический метаболизм миобластов, что может отрицательно сказаться на их способности сливаться во время регенерации мышц in vivо [189].
Поджелудочная железа и жировая ткань
Опосредованное алкоголем нарушение биоэнергетических функций наблюдается при алкогольном панкреатите. Фактически, биоэнергетическая дисфункция постулируется как одна из этиопатологий этого заболевания. На первичной культуре ацинарных клеток поджелудочной железы мышей и человека, а также в линии ацинарных клеток поджелудочной железы AR42J, обработанных этанолом, ацетальдегидом, ЭЭЖК или их комбинациями, было показано, что этанол значительно снижает общую продукцию АТФ и усиливает митохондриальный стресс [190–193]. Ацетальдегид увеличивает количество АТФ, образующегося в результате гликолиза, но ингибирует митохондриальный оборот АТФ, что указывает на компенсаторную метаболическую адаптацию при нарушении OXPHOS. Тем не менее, ЭЭЖК ингибируют как гликолитическую, так и митохондриальную продукцию АТФ; это дает основания полагать, что алкоголь усугубляет неспособность клеток удовлетворять свои энергетические потребности, что приводит к нарушению функции ацинарных клеток и апоптозу. Кроме того, как ацетальдегид, так и ЭЭЖК снижают запасную дыхательную способность и нарушают митохондриальный резерв [190]. Окислительный метаболизм этанола, активирующий MPTP и митохондриальную недостаточность, также предлагается в качестве механизма снижения производства АТФ в ацинарных клетках поджелудочной железы [194, 191]. В частности, было установлено, что этанол или повышение уровня Ca2+, опосредованное ЭЭЖК, служит основной причиной наблюдаемого снижения синтеза АТФ, повышенной активации трипсиногена и гибели клеток, связанной с острым алкогольным панкреатитом [191].
Клинические и доклинические исследования показывают, что алкоголь снижает базальный уровень инсулина в кровотоке [195, 196] и экспрессию циркулирующего инсулина и С-пептида в ответ на глюкозу [197]. Исследования, проведенные группой авторов обзора с помощью теста на толерантность к внутривенной глюкозе с частыми пробами, показали, что ХЗА значительно ухудшает эндокринную реакцию поджелудочной железы на нагрузку глюкозой у ВИО-инфицированных макак [198, 199]. Более того, воздействие алкоголя in vitro снижает секрецию инсулина островками поджелудочной железы человека [200] и грызунов [196, 201, 202] и усиливает апоптоз β-клеток [203–205]. Однако механизмы, ответственные за опосредованное алкоголем нарушение эндокринной функции поджелудочной железы, во многом неясны. При диабете второго типа в β-клетках поджелудочной железы снижается потребление кислорода, отношение АТФ : АДФ и усиливается окислительный стресс. Поскольку функция митохондрий имеет решающее значение для стимулируемого глюкозой выделения инсулина β-клетками, а данные указывают на биоэнергетические нарушения в качестве патомеханизма алкогольного панкреатита, необходимы исследования для определения того, участвуют ли биоэнергетические изменения, опосредованные алкоголем, в механизме наблюдаемого снижения секреции инсулина поджелудочной железой. Сходным образом алкоголь нарушает метаболические функции адипоцитов [206–209], а оптимальная биоэнергетическая функция митохондрий имеет решающее значение для дифференцировки, липогенеза, липолиза и секреции адипокинов [210]. Доклинические и клинические исследования ожирения показывают, что потребление кислорода митохондриями, экспрессия белков OXPHOS и биогенез митохондрий в жировой ткани или изолированных адипоцитах снижаются [211–213]. Однако неизвестно, ухудшает ли алкоголь биоэнергетическую функцию адипоцитов. Таким образом, хотя эти две ткани имеют решающее значение для контроля энергетического метаболизма всего тела, в литературе существует пробел относительно влияния биоэнергетической адаптации, опосредованной алкоголем, на эндокринную функцию поджелудочной железы и жировую ткань.
Головной мозг
Биоэнергетическая адаптация мозга к алкоголю изучалась с целью определения их вклада в развитие зависимости и повреждение нейронов. Исследования на рыбках данио показывают, что острое воздействие алкоголя увеличивает базальное дыхание мозга, КI-опосредованное OXPHOS, эффективность сопряжения, биоэнергетическую эффективность и отношение остаточного потребления кислорода к системе переноса электронов (ОПК/СПЭ). Напротив, хроническое введение алкоголя снижало исходное дыхание, транспорт электронов с участием комплексов I и II, увеличивало состояние ОПК и соотношение ОПК/СПЭ [214]. У мышей, которым вводили алкоголь внутрибрюшинно (3,8 г/кг) и которых выводили из эксперимента через 6 ч (моделирование алкогольного похмелья), наблюдалось снижение малатно-глутаматного дыхания в состоянии 3 и скорости образования АТФ, а также усиление синаптосомного дыхания, вызывающее утечку протонов и резервную дыхательную емкость. Это также сопровождалось снижением активности КI, II, III и IV в синаптосомах, демонстрируя, что индуцированные алкоголем биоэнергетические адаптации в головном мозге потенциально могут быть следствием функциональных изменений митохондрий на уровне синапсов, и что снижение двигательной активности может быть связано с нарушениями мозгового кровообращения [215–217]. Аналогичным образом, через два часа после острой внутрибрюшинной инъекции спирта (50 ммоль/кг) в головном мозге наблюдалось значительное угнетение дыхания в состоянии 3 [218]. Этанол in vitro (50–200 мМ) ингибировал стимуляцию OXPHOS, опосредованную деполяризацией. С точки зрения механизмов было продемонстрировано, что этанол ингибирует потенциалзависимые каналы Ca2+, тем самым препятствуя увеличению концентрации свободного Ca2+ в синаптосомах. Частично это происходило за счет стимуляции митохондриального антипортера Ca2+/Na+, который предотвращает увеличение количества свободного Ca 2+ в митохондриальном матриксе. Предполагается, что ингибирование вызываемой возбуждением стимуляции OXPHOS в синапсах способствует депрессантному и наркотическому действию алкоголя [219]. Спустя восемь часов после прекращения хронического прерывистого воздействия алкоголя митохондрии удлинялись, и в медиальной префронтальной коре наблюдалась картина «митохондрий на нитке». Это было связано со значительным подавлением митохондриальной биоэнергетики, в том числе со снижением работы ЭТЦ, экспрессии гена Mfn2 и усилением деления/фрагментации [37]. В головном мозге не наблюдалось изменений в работе субстратных челноков и окислении этанола, но снижалось отношение НАД+/НАДН и нарушалось OXPHOS [220]. В митохондриях, выделенных из головного мозга мышей, получавших хроническое алкогольное опьянение, была понижена активность КI и V и уровень карнитинпальмитоилтрансфераз 1 (cPT1) и cPT2, которые необходимы для ацилирования жирных кислот от наружной к внутренней митохондриальной мембране для производства АТФ. Это также было связано с уменьшением уровня β-окисления пальмитата; это говорит о том, что нарушение этапа поступления субстрата (функция КI) может влиять на синтез АТФ (функция КV). Кроме того, наблюдалась повышенная утечка цитохрома с, связанная с уменьшением содержания cPT1/cPT2, в то время как снижение количества КI и V происходило параллельно с уменьшением деполяризации потенциала на митохондриальной мембране и продукции АТФ [221]. В целом, значительные доказательные данные подтверждают биоэнергетическую адаптацию нейронов к острому и хроническому воздействию алкоголя.
Изменения биоэнергетики митохондрий наблюдаются и у крысят, чьи матери получали алкоголь во время беременности. В зернистых нейронах мозжечка таких детенышей значительно понижен уровень мРНК митохондриальных генов, кодирующих КII, IV и V. Обработка этанолом клеток мозжечка in vitro снижала экспрессию митохондриальных генов в нейронах, кодирующих КIV и V, нарушала функцию митохондрий и синтез АТФ. Эти данные убедительно свидетельствуют о том, что нарушение функций митохондрий может лежать в основе патофизиологии фетального алкогольного синдрома [222].
В воздействии на биоэнергетическую адаптацию мозга, опосредованную алкоголем, участвуют несколько генов-кандидатов. Было продемонстрировано, что после одной недели приема этанола крысами-подростками агонист рецептора меланокортина 4 (MC4R) уменьшает окислительное повреждение гиппокампа за счет усиления экспрессии двух ключевых митохондриальных генов NRF2 и PGC1α, увеличения объема митохондрий, снижения уровня кальция в митохондриях и увеличения экспрессии белков дыхательных комплексов [223–225]. С помощью парадигмы «пития в темноте» было показано, что вызванное алкоголем окислительное повреждение гиппокампа и активация астроцитов у мышей-подростков были связаны со снижением экспрессии MFN2, что подразумевает нарушение митогорметического ответа и биоэнергетической функции при нейровоспалении, ассоциированном с алкоголем [226].
Таким образом, опубликованные данные свидетельствуют о том, что биоэнергетическая адаптация, опосредованная алкоголем, является общей для всех метаболически активных тканей и приводит к патофизиологическим изменениям (рисунок 6). Как эти биоэнергетические изменения синергетически взаимодействуют с другими патомеханизмами, опосредованными алкоголем, что приводит к повреждению органов-мишеней, еще предстоит полностью понять; такое понимание будет иметь решающее значение для определения терапевтических целей для облегчения бремени болезни.
«Белые пятна» в знаниях об опосредованной алкоголем биоэнергетической адаптации и клеточной функции
Настоящий обзор посвящен описанию опосредованных алкоголем изменений в биоэнергетике тканей, играющих центральную роль в гомеостазе энергетического метаболизма всего организма. Однако хорошо известно, что одним из механизмов хронического злоупотребления алкоголем служит иммунная активация и воспаление, а также неспособность иммунной системы реагировать на инфекции [227–229]. Хотя изменения биоэнергетики тканей, вызванные алкоголем, хорошо охарактеризованы, в литературе недостаточно данных, посвященных опосредованным алкоголем изменениям иммунометаболизма. Ранние работы группы авторов обзора показали, что алкогольная интоксикация нарушает вызванное эндотоксинами увеличение метаболизма глюкозы и подавляет утилизацию глюкозы — особенно тканями, богатыми иммунными клетками [230, 231]. Увеличение скорости метаболизма со стороны клеток иммунной системы является необходимым компонентом адекватной защиты хозяина. Более свежие данные о нарушении иммунометаболизма с участием алкоголя получены в исследованиях альвеолярных макрофагов. Yeligar и его коллеги продемонстрировали, что вызванный этанолом окислительный стресс с участием NOX4 нарушает фагоцитарную функцию этих клеток. Это связывали со понижением уровня базального дыхания, АТФ-связанного дыхания, максимального дыхания и резервной емкости альвеолярных макрофагов, что говорит о биоэнергетической адаптации. Также наблюдалось увеличение гликолитической способности, гликолитического резерва и негликолитического закисления с сопутствующим усилением экспрессии и активности индуцируемого гипоксией фактора 1α (HIF1α), фосфорилированием пируватдегидрогеназы и повышением уровня внеклеточного лактата в альвеолярных макрофагах [232]. Однако иммунные клетки, особенно CD4+ Т-лимфоциты, для удовлетворения энергетических потребностей иммунных реакций полагаются как на гликолиз, так и на OXPHOS [233]. В то время как наивные CD4+ Т-клетки полагаются на β-окисление, при активации они задействуют аэробный гликолиз [233]. Последующая дифференцировка зависит от различных метаболических путей: Th1, Th2 и Th17 — от гликолитического пути, тогда как Treg — от OXPHOS [234, 235]. Результаты недавних исследований авторов обзора впервые показали, что этанол снижает максимальное дыхание, эффективность связывания, продукцию АТФ, связанную с БПК, и уменьшает ИБЗ с одновременным увеличением связанной с БПК утечки протонов в дифференцированных CD4+ T-клетках. Это также было связано с усилением гликолиза, при ингибировании которого предотвращалось опосредованное этанолом увеличение числа клеток Th1 без влияния на Treg. Кроме того, этанол увеличивает объем митохондрий в Treg и изменяет экспрессию генов, участвующих в митофагии. В целом данные свидетельствуют о том, что этанол ухудшает иммунометаболизм CD4+ Т-клеток и процессы восстановления митохондрий, способствуя провоспалительному фенотипу CD4+ Т-лимфоцитов [236]. Вопрос о том, способствует ли такая биоэнергетическая дезадаптация иммунных клеток функциональному изменению адаптивных иммунных реакций у людей, злоупотребляющиз алкоголем, активно изучается.
Заключение и перспективы
Алкоголь и его метаболиты проникают практически во все ткани, вызывая клеточный стресс. Современные методы определения клеточных биоэнергетических измерений и их интеграция с омиксными и таргетными подходами расширили понимание вызываемой алкоголем адаптации для удовлетворения энергетических потребностей в ответ на клеточный стресс. В метаболически активных тканях алкоголь ухудшает биоэнергетические процессы и одновременно усиливает окислительный стресс, снижая метаболическую гибкость. В основе этого лежит нарушение регуляции морфологической и функциональной целостности митохондрий. Такой запускаемый алкоголем порочный круг нарушений биоэнергетики и усиления окислительного стресса потенциально может инициировать клеточное старение и регулируемую гибель клеток, что в конечном итоге приводит к повреждению тканей. Эти биоэнергетические адаптации действуют совместно с другими критическими патофизиологическими механизмами, усугубляющими индуцированное алкоголем повреждение органов-мишеней. В изучении биоэнергетических адаптаций в метаболически активных тканях были достигнуты значительные успехи, но актуальность и значение вклада алкоголя в изменения иммунометаболизма еще предстоит полностью прояснить. Это особенно актуально в связи с иммунным ответом на инфекционное или неинфекционное воздействие, способностью иммунной системы уничтожать бактериальные или вирусные патогены и стойкой реакцией на вакцины. Достижения в фундаментальных знаниях о том, как алкоголь вызывает клеточное повреждение, позволят понять новые стратегии для облегчения бремени болезни.
