
Молекулярные механизмы, общие для преждевременного старения и болезней старости

Аннотация
Старение — основной фактор риска развития многих распространенных заболеваний. Болезни преждевременного старения человека являются перспективными модельными системами для определения и описания клеточных механизмов, лежащих в основе физиологического старения. Также их изучение позволяет лучше понять причины, пусковые факторы и потенциальные стратегии лечения распространенных болезней старости, в том числе неврологических расстройств, диабета, онкологических и сердечно-сосудистых заболеваний. Используя в качестве основной парадигмы преждевременного старения редкую патологию — синдром Хатчинсона-Гилфорда, авторы обсудят общие механизмы преждевременного старения и болезней старости, включая генетические и эпигенетические дефекты, дефекты в метаболических путях; митохондриальный и белковый гомеостаз; клеточный цикл и способность стволовых клеток к регенерации.
Старение — это процесс постепенного функционального истощения на клеточном и организменном уровнях. Болезни, связанные со старением (ageing-associated diseases, AADs), к которым относится большинство патологий сердечно-сосудистой системы, хроническая обструктивная болезнь легких (ХОБЛ), инсульт, болезнь Альцгеймера, хроническая болезнь почек (ХБП) и онкологические заболевания, являются причиной приблизительно половины всех человеческих смертей (см. «Хронические заболевания, связанные со старением») [1,2]. Хотя старение — основной фактор риска развития этих заболеваний, понимание того, как именно оно участвует в их возникновении и развитии, находится в зачаточном состоянии.
Роль старения в развитии болезней человека в основном изучается на животных моделях заболеваний, путем препятствования началу развития и прогрессированию дефектов в тканях, преимущественно вовлеченных в патогенез различных связанных со старением болезней (см. Вставка 1). Несмотря на то, что животные модели являются удобной заменой для изучения основ старения, которые могут быть одинаковы для разных видов, они не идеальны для освещения эффектов старения на человеческие болезни, в силу слишком низкой встречаемости AADs у лабораторных животных в сравнении с людьми [3–5].
Ключевая причина ААDs — снижение функции клеток и тканей, связанное со старением [6,7]. Клеточное старение характеризуется повышением нестабильности генома, изменением метаболизма и потерей регенеративного потенциала. Рассмотрение старения и износа клеток в качестве движущей силы болезней, связанных со старением, позволяет объяснить наблюдаемое при AADs поражение не только ткани, непосредственно вовлеченной в патологический процесс, но и одновременное снижение функции других тканей [6,7]. Функциональный износ тканей многих органов, часто упускаемый из виду при AADs, важен для диагностики и изучения патологии: например, сила рукопожатия и переломы бедра являются индикаторами ХПН и болезни Паркинсона, соответственно [8,9]. Эти наблюдения показывают, что клеточное старение является универсальным принципом, лежащим в основе заболеваний, связанных со старением.
Вставка 1 | Хронические заболевания, связанные со старением
Болезнь Альцгеймера
Хроническое нейродегенеративное заболевание, характеризующееся деменцией, дезориентацией, перепадами настроения, потерей аппетита, сумбурностью речи и неспособностью к координации движений; эта болезнь также ассоциирована с повышенным риском остеопороза и мышечной атрофии. В большинстве случаев семейные формы болезни Альцгеймера обусловлены мутациями в белке-предшественнике β-амилоида (APP) и в пресенилинах 1 и 2, которые усиливают образование β-амилоида — продукта расщепления APP (обычно откладывающегося в сенильных бляшках). Кроме этого, нейрофибриллярные клубки, состоящие в основном из гиперфосфорилированного тау-белка, являются ключевым признаком болезни Альцгеймера.
Атеросклероз
Заболевание сосудов, характеризующееся тем, что артерии становятся менее эластичными и кальцифицируются, вследствие образования холестериновых бляшек, которые препятствуют току крови. Нестабильные бляшки имеют меньшее число гладких миоцитов и более склонны к разрыву, что может приводить к инфаркту или инсульту.
Рак
Группа заболеваний, подразумевающих под собой аномальный рост клеток, что проявляется либо в инвазивной форме (злокачественной), либо неинвазивной (доброкачественной), как результат накопления генетических мутаций, ингибирующих активность генов-супрессоров опухолей, или активацией/оверэкспрессией онкогенов.
Хроническая болезнь почек
Хроническое состояние, определяемое как постепенное снижение функций почек, способное приводить к повышению кровяного давления, анемии, уменьшению костной массы и повреждению нейронов.
Хроническая обструктивная болезнь легких
Группа легочных патологий, при которых уменьшается воздушный поток и затрудняется дыхание, что обусловлено в основном повышением продукции слизи и воспалением (бронхит) либо же разрушением альвеол и расширением воздушных пространств легких (эмфизема). Идиопатический легочный фиброз характеризуется утолщением и рубцеванием легочной ткани, приводящих к ухудшению газообмена между легкими и кровью. Пациенты с ХОБЛ имеют повышенный риск развития болезни Паркинсона.
Сердечная недостаточность
Хроническое состояние, характеризующееся сниженным сердечным выбросом, причиной которого является неспособность сердца адекватно сокращаться вследствие растяжения/истончения стенки желудочков, или же расслабляться из-за их гипертрофии / сниженной эластичности.
Остеопороз
Уменьшение костной массы из-за дисбаланса между процессами остеогенеза и резорбции кости.
Болезнь Паркинсона
Хроническое нейродегенеративное заболевание, при котором нарушается функция моторной системы, что приводит к тремору, ригидности, нарушениям походки и зачастую к деменции и депрессии. Причины наследственных форм болезни Паркинсона включают в себя мутации α-синуклеина, parkin, серин/треониновой богатой лейциновыми повторами протеинкиназы 2 (LRRK2), PTEN-индуцированной киназы 1 (PINK1), DJ1 и TP13A2. Одним из типичных признаков болезни Паркинсона является накопление α-синуклеина в виде телец Леви, которые способствуют гибели клеток в имеющем дофаминергическую иннервацию черном веществе.
Саркопения
Потеря мышечных массы и силы в ходе старения. Пациенты с ХОБЛ, сердечной недостаточностью, раком или ХБП имеют повышенный риск саркопении.
Сахарный диабет 2 типа
Метаболическое расстройства, при котором инсулин не может быть использован должным образом, что частично компенсируется повышением его образования панкреатическими β-клетками. В конце счете последние клетки перестают справляться. Пациенты с СД2 находятся в группе риска развития болезни Альцгеймера и ХБП.
Значительно облегчило изучение старения человека открытие мутаций, лежащих в основе синдромов преждевременного старения (прогероидных синдромов, — прим. ред.) (табл. 1). Наиболее яркими из них являются: синдром преждевременного старения Хатчинсона-Гилфорда (СПХГ) [10–12] и атипичный синдром Вернера, обусловленные дефектами белков ядерной оболочки (равно как и классическая форма синдрома Вернера), синдромы Коккейна, Блума, пигментная ксеродерма, атаксия-телеангиэктазия, трихотиодистрофия, врожденный дискератоз (ВДК), синдром мозаичной смешанной анеуплоидии, причиной которых являются дефекты в системе репарации ДНК, в т.ч. и в обеспечивающих ее белках (табл. 1). Клеточные дефекты, наблюдаемые при этих и других болезнях преждевременного старения, включая нестабильность генома и протеома, изменение метаболизма и потерю регенеративного потенциала, совпадают с таковыми при физиологическом старении организма. Кроме того, существуют поразительные сходства между дефектами организма при некоторых прогероидных синдромах и AADs. Тем не менее, т.к. синдромы преждевременного старения отображают лишь некоторые аспекты физиологического старения человека, неудивительно, что прогероидные патологии имеют общее лишь с некоторыми из AADs (табл. 1). К примеру, при СПХГ наблюдаются значительные нарушения сердечно-сосудистой системы и остеопороз, в то время как болезни преждевременного старения, связанные с дефектами белков репарации ДНК, характеризуются предрасположенностью к онкологическим и нейродегенеративным заболеваниям. Наличие таких параллелей предполагает общность этиологии у прогероидных синдромов и AADs.
Таблица 1 | Прогероидные синдромы
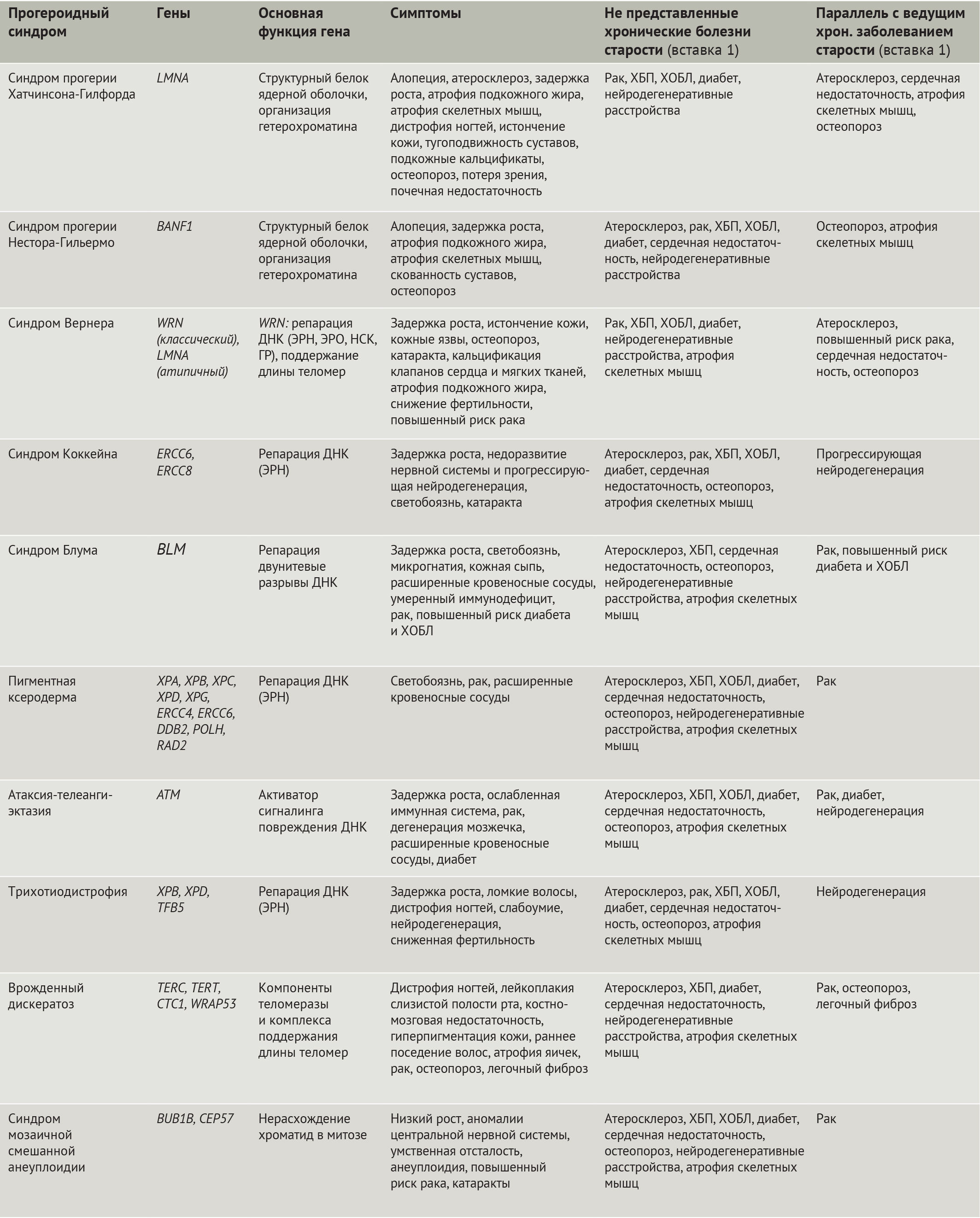
В данном обзоре авторы обсудят дефекты в клеточных и молекулярных механизмах, общих для болезней преждевременного старения и для физиологического старения, а также подробно осветят роли этих путей в развитии некоторых болезней, ассоциированных со старением. В качестве парадигмы взаимосвязи между преждевременным и физиологическим старением и AADs авторы рассмотрят СПХГ, в силу того, что это — одна из наиболее полно изученных болезней преждевременного старения, демонстрирующая широкий спектр дефектов на клеточном, тканевом и организменном уровнях, общих с AADs.
СПХГ в роли Розеттского камня для механизмов старения
СПХГ — редчайшая болезнь преждевременного старения с частотой встречаемости 1 случай на 4–8 млн новорожденных [13]. Клинические особенности СПХГ проявляются уже вскоре после рождения и включают: тотальную алопецию, атрофию подкожно-жировой клетчатки и скелетных мышц, ониходистрофию, тугоподвижность суставов, появление морщин и подкожных кальцификатов, нарушения структуры костей и потерю зрения (табл. 1) [13]. Болезнь неминуемо приводит к гибели в возрасте 15–17 лет, при этом основными причинами смерти являются атеросклероз-подобная прогрессирующая патология сердечно-сосудистой системы, инфаркт миокарда и инсульт [14]. СПХГ в основном обусловлен гетерозиготной сайленс-мутацией (G608G) в гене LMNA, приводящей к синтезу прогерина — структурного ядерного белка ламина А, аномальной формы [10,11,15].
Ламин А дикого типа претерпевает посттрансляционный процессинг: при отщеплении С-конца под действием гомолога CAAX-пренил-протеазы 1 (кодируемой геном ZMPSTE24, также известным у мышей как ZMPSTE24) образуется зрелый ламин А, молекулы которого организуются в промежуточные филаменты, далее встраивающиеся в ядерные ламину и матрикс (рис. 1) [16,17]. При СПХГ мутация 1824С>T (LMNAG608G) активирует криптический (скрытый, — прим. ред.) сайт сплайсинга, что приводит к удалению 50-аминокислотной последовательности, содержащей сайт узнавания ZMPSTE24. В результате этого прогерин накапливается на периферии ядра и вызывает нарушения механохимических свойств ламины, что можно понять по аномальной форме ядра, наблюдаемой в клетках у пациентов с СПХГ (рис. 1). Подобные клеточные дефекты обусловлены мутациями LMNA, вызывающими атипичные формы СПХГ (включая LMNAG608S и LMNAE145K) [16,18]. Нарушения механических свойств и механотрансдукции ядерной ламины, как считается, играют роль в патологии у пациентов с СПХГ, т. к. многие поражаемые ткани, такие как сосуды, кости и суставы, особенно подвержены механическим воздействиям [17,19].
СПХГ отличается от других прогероидных синдромов своим ранним началом, выраженностью признаков старения и большим числом вовлеченных в патологический процесс тканей (табл. 1) [15,20]. В отличие от СПХГ, классическая форма синдрома Вернера (прогерии взрослых) проявляется на третьем десятилетии жизни остеопорозом и онкологическими заболеваниями.
Раннее проявление дефектов старения у больных СПХГ, вероятно, отчасти связано с доминантно-негативным действием прогерина, в то время как другие синдромы преждевременного старения зачастую ассоциированы с рецессивными мутациями, приводящими к утрате функции белков репарации ДНК, что может быть частично скомпенсировано альтернативными путями репарации [20]. Как следствие, у больных СПХГ наблюдается широкий спектр тканевых дефектов (от остеопороза до связанных со старением патологий скелетных мышц и сердечно-сосудистой системы). В свою очередь прогероидные синдромы, вызванные нарушением репарации ДНК, в своей массе ассоциированы с высокой предрасположенностью к онкозаболеваниям и, в некоторых случаях, с прогрессирующей нейродегенерацией (табл. 1) [20]. Любопытно, что по непонятным причинам у пациентов с СПХГ не повышен риск развития диабета и ХБП. Кроме того, у них не происходит и нейродегенерации; вероятно, причина этого — микроРНК-9 (miR-9), которая подавляет экспрессию ламина А и прогерина в нейронах. Также несмотря на значительные повреждения ДНК, у пациентов с СПХГ не развиваются опухоли в детском возрасте, что может быть связано с изменением структуры внеклеточного матрикса, препятствующим инвазии опухоль-инициирующих клеток [21,22], и с тем, что прогерин оказывает протективный эффект в отношении опухолевого роста [23].
Несмотря на то, что некоторые симптомы заболеваний, связанных с возрастом не наблюдаются при СПХГ (табл. 1), многие ключевые признаки клеточного старения, отмечаемые в тканях, поражаемых при AADs, встречаются и у пациентов с СПХГ (рис. 1) [24]. Это яркое проявление дефектов старения при СПХГ было объяснено негативным действием прогерина на ядерную ламину, являющейся основным каркасом клеточного ядра млекопитающих [25]. Установлено, что дефекты ламины участвуют в механизме возникновения большинства признаков, общих для старения и AADs (рис. 1), включая утрату генетической и эпигенетической целостности, укорочение теломер, нарушение гомеостаза белков (протеостаза), перепрограммирование метаболизма, митохондриальную дисфункцию, клеточное старение, нарушение поддержания функции стволовых клеток и их регенеративного потенциала [26]. Таким образом, экспрессия прогерина вовлечена в различные механизмы, в то время как при прогероидных синдромах Вернера, Блума и Коккейна наблюдается более ограниченный спектр дефектов, обусловленных в основном нарушением репарации ДНК [27–34]. Важность ядерной ламины в старении иллюстрируется еще одной болезнью преждевременного старения — синдромом прогерии Нестора-Гильермо (СПНГ), причиной которого служит гомозиготная мутация в гене белка, локализованном в ядерной ламине, — фактора барьера аутоинтеграции 1 (BANF1) [35]. Подобно СПХГ, при СПНГ наблюдаются тугоподвижность суставов, дистрофия подкожно-жировой клетчатки и скелетных мышц и отставание в росте (рис. 1) [35]. Причастность дисфункции ядерной ламины к различным признакам клеточного старения дает возможность рассматривать СПХГ как подходящую модель для обсуждения параллелей между преждевременным старением и AADs на клеточном и молекулярном уровнях.
СПХГ имеет особенное отношение к обычному старению, потому как у физиологически стареющих людей также вырабатывается небольшое количество прогерина — благодаря периодически происходящему самопроизвольному открытию криптического сайта сплайсинга, который активирован у пациентов с СПХГ [28, 36]. В силу доминантно-негативного действия прогерина, его небольшие количества, вероятно, участвуют в процессе нормального старения [36]. Эта точка зрения подтверждается высокой схожестью клеточных и организменных дефектов у пациентов с СПХГ и физиологически стареющих индивидов [20,26]. Вдобавок к этому, устранение экспрессии прогерина и ламина А на генетическом уровне увеличивает продолжительность жизни мышей [37], а мутация 1968G>A в гене LMNA, приводящая к более слабой активации скрытого сайта сплайсинга прогерина, своим следствием имеет менее яркую выраженность связанных со старением патологий [38].
Единственный, к тому же достоверно определенный, генетический дефект у пациентов с СПХГ, прямая связь между прогерином и многочисленными признаками клеточного старения, экспрессия патогенного белка в ходе физиологического старения и манифестация связанных со старением патологий во многих тканях — все это делает СПХГ привлекательной моделью для изучения роли механизмов старения в патогенезе AADs.
Нестабильность генома при старении и AADs
Человеческий геном постоянно подвергается влиянию факторов, повреждающих ДНК и угрожающих клеточному гомеостазу. Этому противодействуют механизмы, исправляющие повреждения и не дающие укорачиваться концам хромосом. Снижение их эффективности с возрастом и при AADs приводит к износу клетки [39–42].
Дефекты в механизмах репарации ДНК
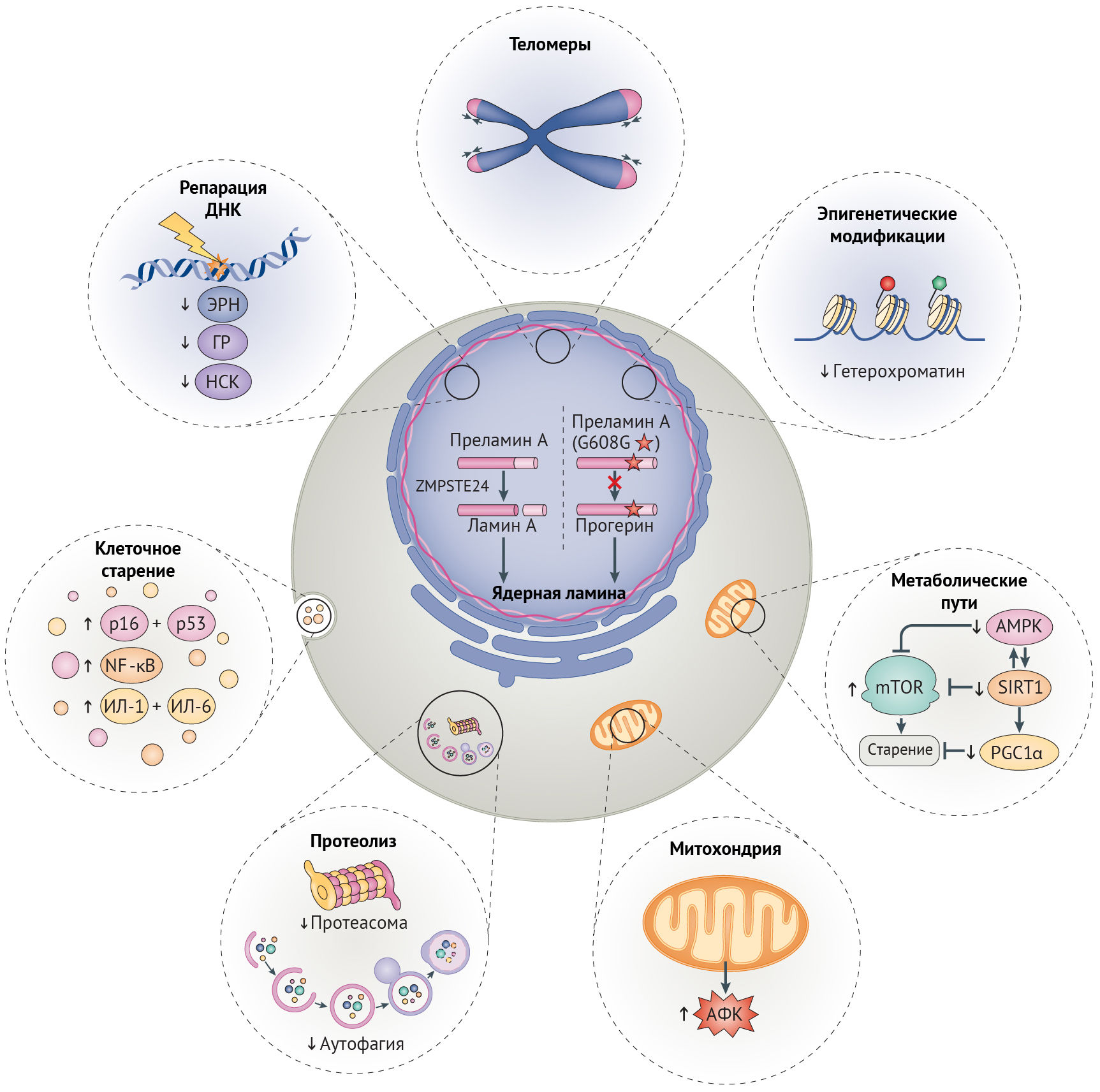
Целостность генома поддерживают несколько механизмов репарации ДНК. Механизм эксцизионной репарации нуклеотидов (NER, ЭРН) исправляет повреждения на одной цепи ДНК [40–42], в то время как двунитевые разрывы (DSBs, ДНР), приводящие к крупным хромосомным перестройкам и угрожающие выживанию клетки, в первую очередь и с высокой точностью исправляются с помощью гомологичной рекомбинации, но могут репарироваться и методом негомологичного соединения концов (NHEJ, НСК), подверженному ошибкам. Эффективность этих механизмов репарации снижается в процессе старения (рис. 2) [39–42], результатом чего служит повышение с возрастом количества хромосомных аберраций и перманентной активации сигналинга повреждений ДНК [43].
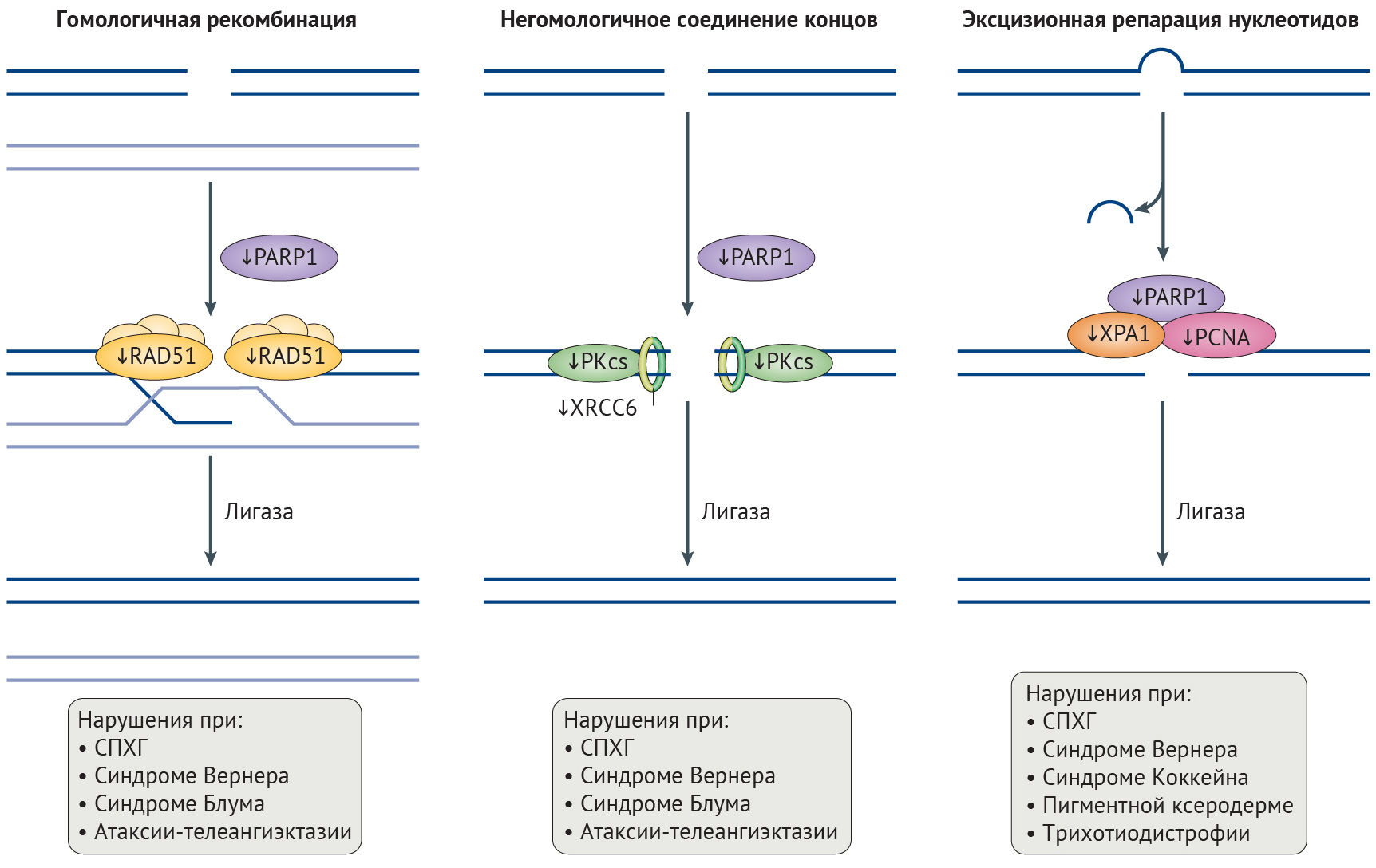
PCNA, ядерный антиген пролифелирующих клеток.
Неполноценность репарации ДНК при СПХГ. Хроническая активация сигналинга повреждений ДНК при СПХГ приводит к повышению числа ДНР и неполноценному их исправлению [44]. Дефекты репликации, включая коллапс репликативной вилки, играют большую роль в появлении ДНР и особенно часто наблюдаются при СПХГ. Причиной этого служат уменьшение содержания одного из необходимых для репликации ДНК белков скользящего зажима — ядерного антигена пролиферирующих клеток (PCNA), а также нарушение взаимодействий между ним и ламином А и повышение активности протеолитической деградации фактора репликации C1 (рис. 2) [45–47]. Эффективность исправления ДНР, осуществляемого с помощью механизмов гомологичной рекомбинации и НСК, снижена в силу замедленного рекрутинга белков репарации (таких как TP53-связывающий белок 1 (TP53BP1) и RAD51), ингибирования белка репарации ДНК поли(АДФ-рибоза)-полимеразы 1 (PARP1), а также пониженного содержания участвующих в НСК белков — DNA-PKcs и перекрестно-комплементирующего белка репарации рентгеновских повреждений (XRCC6) [30,48,49]. Более того, некорректное связывание белка репарации XPA в участках ДНР также препятствует репарации и уменьшает эффективность ЭРН (рис. 2) [45].
Неполноценность репарации ДНК при других прогероидных синдромах. О роли ЭРН и репарации ДНР в процессе старения и патогенезе AADs убедительно говорят прогероидные фенотипы при синдроме Коккейна, пигментной ксеродерме, трихотиодистрофии (вызываемых дефектами белков ЭРН) и атаксии-телеангиэктазии, синдромах Вернера и Блума (вызываемых нарушением репарации ДНР) (рис. 2; табл. 1) [20]. Высокий процент патологий нервной системы среди пациентов с некоторыми из этих прогероидных синдромов говорит о том, что жизнедеятельность постмитотических нейронов существенно зависит от эффективности репарации ДНК, что, вероятно, связано с накоплением повреждений ДНК в течение жизни организма, причиной чего являются высокая транскрипционная активность и воздействие оксидативного стресса [50]. Удивительно, но эффективность системы репарации ДНК в головном мозге низка, и становится еще ниже при нейродегенеративных заболеваниях [50]. Это наблюдение согласуется с повышенной встречаемостью анеуплоидных нейронов при болезни Альцгеймера [50,51], вероятно, связанной с дефектами НСК, что хорошо соотносится с данными о прямом ингибировании ЭРН мутантным белком parkin при наследственной форме болезни Паркинсона (см. Вставка 1) [52]. Более того, при обеих данных патологиях фибробласты и лимфобласты обладают повышенной чувствительностью к вызванным рентгеновским излучением повреждениям ДНК [50,53]. Увеличенный уровень повреждения ДНК может быть причиной как функциональных нарушений в нейронах, так и уменьшения числа нервных клеток вследствие индукции апоптоза [50].
Неполноценность репарации ДНК при AADs. Повышение уровня повреждений ДНК наблюдается и в некоторых специфических типах клеток, поражаемых при AADs. В гладких миоцитах сосудов (ГМС), повреждаемых при атеросклерозе, уровень повреждений ДНК при воздействии активных форм кислорода (АФК) выше, вследствие возрастания механического стресса, накопления липопротеинов и воспалительного ответа. Последующее снижение пролиферации и апоптоз ГМС может привести к разрыву атеросклеротической бляшки [54]. ГМС очень быстро «стареют» у пациентов с СПХГ, что сочетается с дефектами гомологичной рекомбинации [48]. Подобно этому, лимфоциты у больных диабетом второго типа (СД2) обладают сниженной способностью исправлять повреждения ДНК, вызванные АФК, а β-клетки поджелудочной железы страдают из-за недостаточности системы НСК [55,56]. Более того, дифференцировка образующих кость остеобластов нарушается при повреждениях ДНК, что является причиной хрупкости костей у мышиных моделей прогерии [57]. Согласно этим данным логично предположить, что дефектное НСК может приводить к остеопении, менее тяжелой старческой форме остеопороза [58].
Пониженная эффективность репарации ДНК ведет к накоплению геномных мутаций в ходе старения, что способствует опухолеобразованию благодаря активации онкогенов или инактивации генов-супрессоров опухоли. Действительность этого механизма подтверждается высокой встречаемостью опухолей при большинстве прогероидных синдромов с нарушениями репарации ДНК (табл. 1) [20].
Примеры выше наводят на мысль, что повреждения ДНК являются ключевым фактором развития AADs, но главный вопрос остается открытым: стохастически ли влияет репарация ДНК на целостность генома, или же она затрагивает специфические участки последнего [59].
Укорочение теломер
Теломеры — повторяющиеся последовательности на концах хромосом, прикрытые шелтериновым комплексом, включающим в себя факторы, которые связываются с повторами теломер 1 и 2 (TERF1 и TERF2), и препятствующим распознаванию концов хромосом в качестве ДНР [60]. Теломеры укорачиваются в ходе деления клетки (процесс, известный как истощение теломер) и при достижении критического размера могут активировать сигнальные каскады повреждения ДНК и запускать старение клетки. Теломераза — комплекс, состоящий из обратной транскриптазы (TERT) и теломеразного РНК-компонента (TERC), который удлиняет теломеры при каждом клеточном цикле. Она активно экспрессируется в стволовых клетках эмбриона, но в то же время не обнаруживается в большинстве других человеческих клеток [61].
У пациентов с СПХГ определяется повышенная активация сигналинга о повреждении ДНК; их теломеры короче [62, 63], а число хромосомных аберраций — выше из-за дефектной гомологичной рекомбинации [64]. Подобно этому у прогероидных мышиных моделей с нокаутом гена Terc наблюдается соединение хромосом конец в конец [20]. Прогерин в свою очередь может усиливать повреждения теломер, нарушая ламин А и TERF2-зависимые системы стабилизации концов теломер, что и происходит у пациентов с атипичным синдромом Вернера [65–68].
Повреждения теломер могут запускать синтез прогерина в клетках дикого типа, а потеря функций теломеразного комплекса лежит в основе прогероидного синдрома ВДК, исходя из чего можно предположить, что укорочение теломер имеет определяющую роль в развитии AADs (табл. 1) [20,69]. Генетическая абляция РНК Terc у мышей приводит к укорочению теломер до критической длины спустя несколько поколений, следствием чего является развитие гипертрофии кардиомиоцитов, снижение функции левого желудочка и повышение систолического артериального давления, характерные для физиологического старения [61]. Кроме того, у мышей с нокаутом Terf2 наблюдается усиленный апоптоз кардиомиоцитов [60]. Роль укорочения теломер в патогенезе заболеваний сердечно-сосудистой системы подтверждается и атеросклеротическим фенотипом у мышей с нокаутом Terc, а также наблюдением, что и у человека эндотелиоциты и ГМС в регионах, подвергающихся повышенному гемодинамическому стрессу, и в атеросклеротических областях имеют более короткие теломеры [60]. Повышенная активность теломеразы может быть как полезна, т. к. повышается срок жизни эндотелиоцитов, так и вредна, потому что способствует пролиферации лейкоцитов и образованию в интиме новых ГМС, тем самым обостряя течение атеросклероза [60].
Истощение теломер также является причиной остеопороза у пациентов с ВДК. Теломеры в хондроцитах и лейкоцитах периферической крови у пожилых больных остеопорозом обладают меньшей длиной, а восстановление активности теломеразы в остеобластах усиливает образование костной ткани [70]. Мутации, приводящие к потере функции TERT и истощению теломер, обуславливают развитие и старческих эмфиземы и фиброза легких [71]. Исследования на мышиных моделях дают возможность предположить, что укорочение теломер ускоряет развитие старческой эмфиземы благодаря уменьшению способности легких противостоять стрессу, индуцированному токсинами [72,73]. Было обнаружено, что при ХБП истощение теломер снижает жизнеспособность клеток и усиливает повреждение почек в ходе ишемии-реперфузии [71]. Вполне возможно, это действительно и для β-клеток поджелудочной железы, выживаемость и функциональность которых снижена при высоком уровне сахара в крови у мышей с нокаутом Tert [71].
Хотя экспрессия TERT в фибробластах у больных СПХГ и может нивелировать некоторые клеточные дефекты, связанные со старением, наблюдение, что у экспрессирующих TERT и прогерин человеческих фибробластов тем не менее проявляются некоторые дефекты старения, позволяет говорить о наличии и других механизмов, задействованных в клеточном старении — регулирующих целостность теломер и других участков генома или запускающих старение [32,74].
Эпигенетические дефекты
Регуляция структуры хроматина достигается с помощью метилирования ДНК и посттрансляционных модификаций гистонов, что влияет на целостность генома, экспрессию генов и в конечном счете на здоровье клетки и развитие заболеваний [26,75].
Изменения в метилировании гистонов при старении
В клетках стареющих лиц наблюдается снижение содержания гистонов в целом и постепенное снижение активности триметилирования лизина в 9 и 27 положениях гистона H3 (H3K9me3 и H3K27me3) — репрессивных меток, способствующих компактизации хроматина (рис. 3) [26]. Утрата гетерохроматина усиливается при характерном для старения ингибировании α-гомолога белка гетерохроматина 1 (HP1α, также известного как CBX5), комплекса ремоделирования и деацетилирования нуклеосом (NuRD) и белков группы Polycomb (PCG), каждый из которых является эпигенетическим сайленсером [20,32,76]. Уменьшение доли гетерохроматина, белков-участников комплекса NuRD и HP1α было отмечено и в клетках пациентов с СПХГ [28, 77]. Потеря комплекса NuRD и сниженная экспрессия H3K27-специфичной метилтрансферазы EZH2 в этих клетках приводит к потере репрессивной метки H3K27me3 гетерохроматина (рис. 2) [32,77]. Любопытно, что нокдаун H3K27-деметилазы UTX1 у Caenorhabditis elegans (один из видов свободноживущих нематод, — прим. ред.) увеличивает продолжительность жизни последних на 30 % [78]. Сниженная активность EZH2, компонента ингибиторного комплекса Polycomb 2 (PRC2), играет роль в развитии СД2, поскольку кондиционная делеция EZH2 в панкреатических β-клетках молодых мышей индуцирует клеточное старение, что приводит к уменьшению количества и пролиферации этих клеток и тем самым обуславливает диабетический фенотип [79].
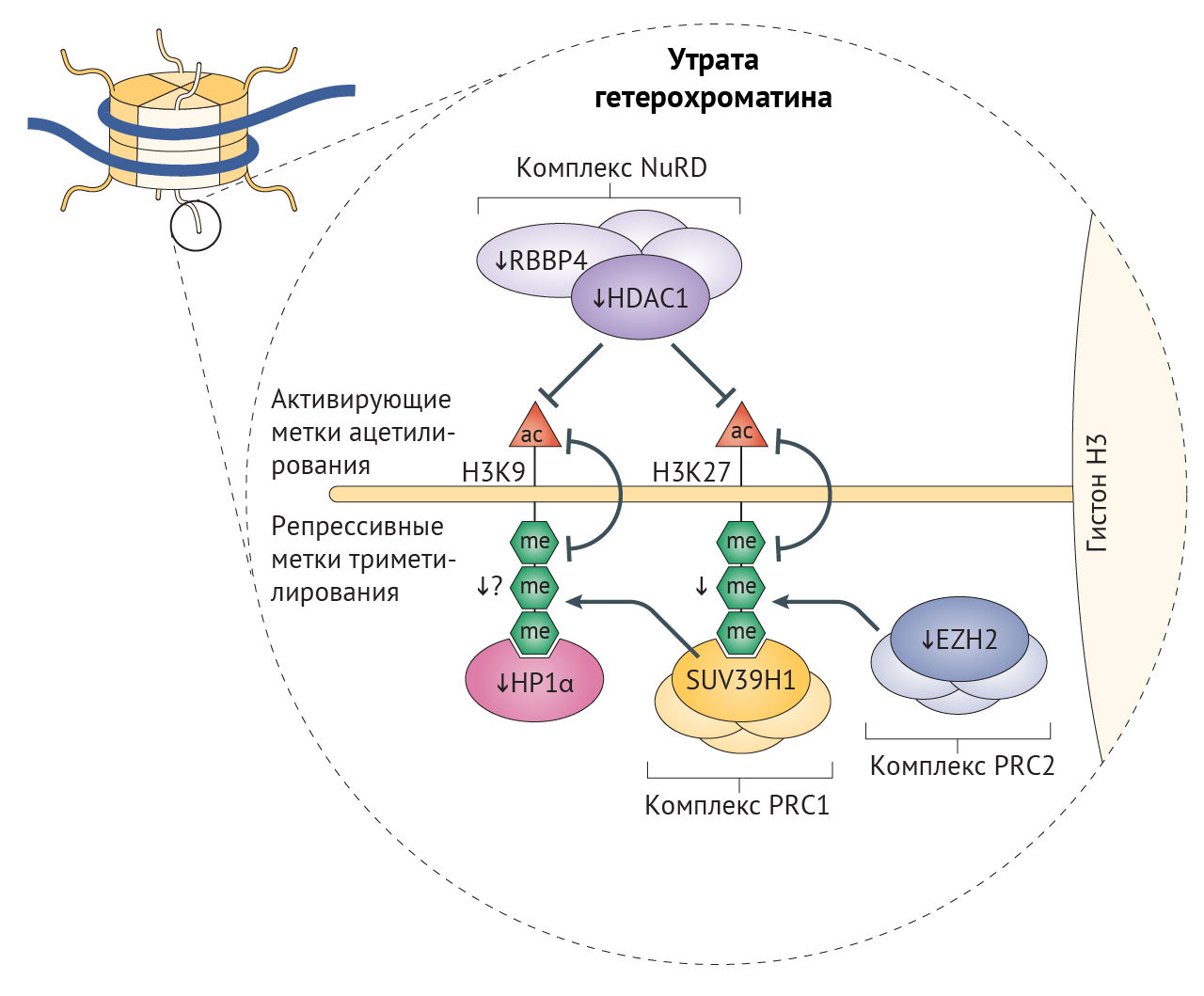
Главная функция комплекса PRC2 — стабилизация H3K27me3-меток, с которыми связывается PRC1-комплекс, и индукция транскрипционного сайленсинга в результате стимуляции метилтрансферазы лизина гистонов SUV39H1, обеспечивающей триметилирование лизина H3 в девятом положении (рис. 3) [80]. Нарушения в ядерной ламине влияют на локализацию белков PCG, что приводит к глобальной потере H3K9me3 при СПХГ, а она, вероятно, является причиной дефектов механизмов поддержания длины теломер и аберрантной активации перицентромерных сателлитных повторов, «молчащих» в норме [32,77,81]. Постепенная утрата HP1α, связывающегося с H3K9me3, может также способствовать снижению доли гетерохроматина, что не удается компенсировать с помощью повышения содержания гетерохроматиновой метки H4K20me3 [77]. Постепенная потеря H3K9me3, ведущая к недостатку белка Вернера в человеческих клетках, вероятно, является результатом уменьшения активности SUV39H1 и связывания HP1α [82]. В то же время у мышиной модели прогерия-подобного фенотипа с делецией фермента ZMPSTE24, осуществляющего процессинг преламина А, наблюдалось повышение H3K9me3. У этой модели утрата SUV39H1 приводила к восстановлению целостности генома и увеличивала срок жизни [83]. Снижение метилирования H3K9 также связано с некоторыми AADs, а при ХПБ вызывает изменения экспрессии генов, характерные для сосудистого воспаления [84]. При семейной болезни Альцгеймера из-за накопления мутантного тау-белка снижаются уровни H3K9me2 и HP1α, результатом чего является повышение экспресии нейротоксичного piwi-подобного белка 1 (PIWIL1) [85]. Более того, снижение экспрессии HP1α встречается во многих злокачественных опухолях, коррелируя с неблагоприятностью прогноза [86].
Старение влияет на ацетилирование гистонов
Массовое снижение метилирования гистонов при СПХГ сопровождается гипоацетилированием гистонов H2B и H4, обусловленным, возможно, ослаблением связи гистон-ацетилтрансферазы KAT8 с ядерной ламиной [87]. Примечательно, что как оверрэкспрессия KAT8, так и лечение ингибиторами гистон-деацетилазы (HDAC) продлевают жизнь нокаутированным по Zmpste24 мышам с прогерия-подобным фенотипом [87]. Метка ацетилирования H4K16 (H4K16Ac), связанная с активацией транскрипции и ее репрессией, способствует НСК и гомологичной рекомбинации; а повышение содержания метки продлевает срок жизни клетки [88, 89]. Терапия ингибиторами HDAC способна предотвратить связанное со старением ухудшение когнитивных функций и уменьшить тяжесть ишемического инсульта, болезни Паркинсона и остеопороза у мышиных моделей благодаря изменению экспрессии генов, способствующих и препятствующих болезни [90,91]. HDACs, включая сиртуины, регулируют ацетилирование и негистоновых белков, в число которых входят мастер-регуляторы клеточного роста и метаболизма, на основании чего можно сделать вывод о непосредственной роли эпигенетической регуляции метаболического контроля старения [26].
Клеточный метаболизм
Метаболический сигналинг критически важен для поддержания клеточного гомеостаза, т. к. благодаря ему распределяется энергия в пользу необходимых защитных механизмов, стоящих на страже целостности генома, эпигенома, протеома и органелл. Его дизрегуляция, запускающая каскады разрушительных событий в клетке, оказывает большое влияние на патогенез AADs.
Нутриент-чувствительные метаболические пути
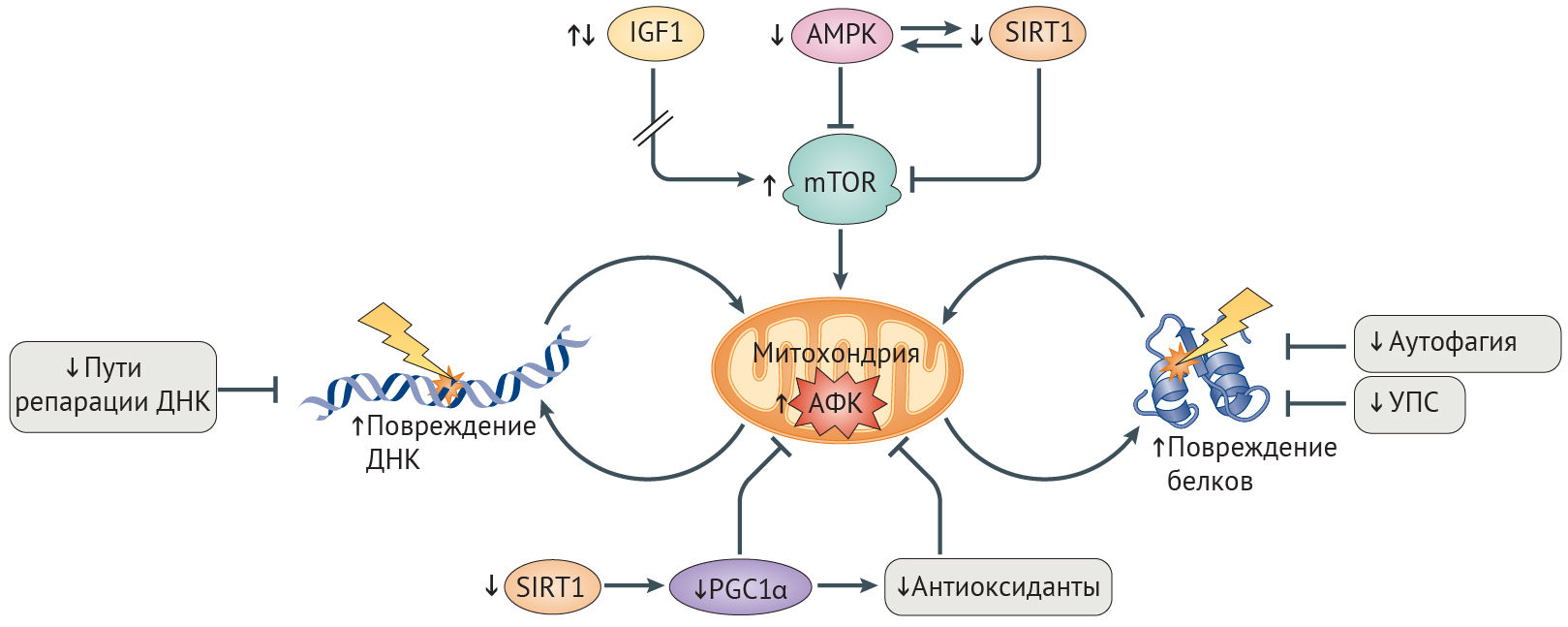
Обеспечение надлежащего баланса между катаболическими и анаболическими путями необходимо для поддержания адекватного уровня энергии в клетке. Нарушение этого равновесия вызывает сбои в клеточном гомеостазе и ведет к старению клетки и развитию AADs [26]. Нутриент-чувствительные сигнальные пути, включая пути инсулино-подобного фактора роста 1 (ИФР-1) и инсулина (IIS), а также сиртуина 1 (SIRT1) и АМФ-активируемой протеинкиназы (АМФК), регулируют метаболический статус клеток и играют заметную роль в физиологическом старении [26].
Дизрегуляция сигналинга ИФР-1 является причиной старения. Сигнальный путь IIS активируется в условиях избытка нутриентов, к коим относятся инсулин, ИФР-1 и свободные аминокислоты. Это приводит к активации пути mTOR (рис. 4), а различные эффекторы на последующих этапах каскада опосредуют повышение синтеза белка и активируют другие анаболические процессы [92]. Снижение IIS-сигналинга, обусловленное генетическим полиморфизмом или же недостатком калорий, способствует долголетию и здоровью [26]. Тем не менее эти эффекты зависят от возраста организма, длительности и степени репрессии IIS, а также от интенсивности клеточных стрессоров, таких как воспаление. В то время как кратковременная и умеренная репрессия IIS может оказаться полезной, подавляя клеточный рост ради перераспределения энергии в пользу восстановления клеточных повреждений, долговременная репрессия —- губительна и способствует старению [92]. У мышей с прогерия-подобным фенотипом, нокаутированных по Zmpste24, уровень ИФР-1 резко понижен в течение всей жизни, а терапия ИФР-1 увеличивает продолжительность жизни и отсрочивает проявление признаков прогерии [93]. Болезнь Паркинсона характеризуется длительным ингибированием mTOR, способствующим смерти нейронов; активация же mTOR у животных моделей позволяет частично предотвратить этот процесс [94].
Долговременная активация IIS также может оказывать негативный эффект на здоровье клетки (рис. 4). СД2-индуцированная хроническая активация mTOR инактивирует субстрат инсулинового рецептора 1 (IRS1) с помощью петли обратной связи, отключающей инсулин и рецепторы к ИФР-1 от дальнейшего сигналинга и вызывающей тем самым инсулинорезистентность [94]. Этот механизм лежит в основе возникновения связанных с диабетом симптомов у больных с синдромом Вернера, тяжесть которых может быть уменьшена ингибиторами mTOR [20,95]. Нейроны обладают высокой чувствительностью к изменениям в IIS-сигналинге в силу высокого уровня метаболизма в них и зависимости от глюкозы [94]. У пациентов, страдающих от болезни Альцгеймера, симптомы диабета (причиной которых служат разобщение и неэффективность передачи сигнала рецептором ИФР-1, и которые обостряются при ингибировании инсулиновых рецепторов β-амилоидами) предшествуют когнитивным нарушениям за десятилетия. Лечение рапамицином, ингибитором mTOR, отчасти приводит в норму сигнальный путь инсулин-mTOR и уменьшает дефекты в нейронах животных моделей болезни Альцгеймера [94,96].
Окислительное фосфорилирование (OXPHOS). Процесс перемещения электронов от доноров электронов к акцепторам, и запасание выделяющейся при этом движении энергии в форме АТФ. У эукариот происходит на внутренней митохондриальной мембране в участках расположения цепи переноса электронов.
Снижение активности сиртуина и АМФК способствует развитию AADs. В условиях стресса и повышенного содержание НАД+ деацетилаза белков SIRT1 активирует метаболические пути, которые повышают уровень энергии в клетке и способствуют ее выживанию [26]. В ходе старения повышенная активность сигналинга повреждений ДНК истощает уровень НАД+, снижая активность SIRT1 (рис. 4) [97]. Важность снижения активности SIRT1 при старении подтверждается наблюдением, что ее восстановление у мышей с прогерия-подобным фенотипом и нокаутом Zmpste24 в результате терапии активатором SIRT1, ресвератролом, приводит к снижению выраженности остеопоротических изменений и увеличению продолжительности жизни [98]. Стимуляция SIRT1 также приводит к облегчению симптоматики болезни Альцгеймера, т. к. при этом ингибируется mTOR, чрезмерно активированный при данной патологии, и индуцируются mTOR-независимые расщепление амилоида и деградация тау-белка (табл. 1) [92].
Повышение активности SIRT1 увеличивает продолжительность здоровой жизни посредством активации АМФК. В ответ на повышение отношения АМФ/АТФ АМФК ингибирует mTOR, стимулирует катаболизм липидов и глюконеогенез и по механизму положительной обратной связи активирует SIRT1 (рис. 4) [99]. Аналогично SIRT1 активность АМФК снижается с возрастом. Соответственно, лечение активатором АМФК, метформином, увеличивает продолжительность жизни C. elegans и мышей, уменьшает выраженность фиброза почек у больных ХБП и оказывает антидиабетический эффект [26, 100]. Метаболические дефекты при СД2 также уменьшаются благодаря усиленной активации SIRT6, возможно путем подавления ИФР-1-сигналинга [92]. У пациентов с СПХГ снижено содержание SIRT6, а оверэкспрессия этого белка несколько уменьшает выраженность старческого фенотипа фибробластов у пациентов с СПХГ и увеличивает срок жизни мышей дикого типа [101,102].
Хотя связь между старением и IIS, АМФК и сиртуин-опосредованным сигналингом очевидна, но то, как именно данные активаторы сигнальных путей влияют на клеточное старение при патологических процессах, известно лишь отчасти. В этом контексте важным фактором, влияющим на клеточное старение, является метаболический контроль целостности митохондрий.
Митохондриальная дисфункция и оксидативный стресс
Митохондрии производят АТФ кислород-зависимым методом окислительного фосфорилирования (OXPHOS), путем образования протонного градиента по обе стороны внутренней митохондриальной мембраны (с помощью белковых комплексов цепи переноса электронов), далее используемого АТФ-синтазой. В дополнение к этому, митохондрии обеспечивают регуляцию глюконеогенеза, окисления жирных кислот, уровня внутриклеткочного кальция и апоптоза [103].
Снижение активности OXPHOS при СПХГ, физиологическом старении и AADs. В ходе старения целостность митохондрий нарушается, что проявляется снижением трансмембранного потенциала, повышением образования АФК в процессе синтеза АТФ, дисрегуляцией гомеостаза кальция, индукцией апоптоза и повышением числа мутаций в митохондриальной ДНК (мтДНК, кодирующей белки-участники OXPHOS) [103]. Связь между целостностью митохондрий и старением продемонстрирована с помощью нокина мтДНК-полимеразы POLGα у мышей, которая в результате проявляла сниженную способность к коррекции ошибок репликации. У этих мышей эффективность OXPHOS была уменьшена вследствие накопления мутаций в мтДНК, что являлось причиной ускоренного старения [104]. При СПХГ наблюдается аномальное набухание и фрагментация митохондрий [105], а активность OXPHOS начинает снижаться с раннего возраста [106]. У мышей с нуль-мутацией Wrn митохондрии увеличены, а количество мутаций мтДНК и образование АФК в процессе генерации АТФ повышены [107].
Система OXPHOS является основным поставщиком АФК [108]. Длительное повышение уровня АФК ведет к повреждению клеточных макромолекулярных структур, включая белки комплексов OXPHOS и мтДНК, создавая порочный круг, в котором АФК нарушают целостность митохондрий, что приводит к дальнейшему нарастанию оксидативного стресса (рис. 4) [109]. При болезни Альцгеймера повышенные уровни АФК предшествуют и способствуют образованию β-амилоидных и тау-содержащих бляшек и нейрофибриллярных клубков [103], которые все больше ухудшают работу OXPHOS-комплексов [110]. Подобная самоподдерживающаяся петля наблюдается и при семейной болезни Паркинсона, при которой патогенные белки усиливают образование АФК, снижают эффективность OXPHOS и в конечном счете запускают гибель нейронов [103]. Генетическое выключение TFAM, фактора выживания мтДНК, схожим образом приводит к болезни Паркинсона у мышей [103]. При атеросклерозе постоянное повышение АФК способствует образованию бляшек посредством ингибирования OXPHOS и окисления липопротеинов низкой плотности, которые оказывают свое повреждающее действие, привлекая моноциты [103]. Установлено, что митохондриальная дисфункция индуцирует воспалительные реакции при ХОБЛ [111]. Нарушение целостности митохондрий, вызванное АФК, далее способствует кальцификации сосудов, что наблюдается у лиц с СПХГ, атеросклерозом, остеопорозом, ХБП, СД2 и болезнью Альцгеймера [112,113].
Снижение эффективности антиоксидантной системы и митохондриального биогенеза при СПХГ и AADs. Клетки могут бороться с порочным взаимодействием между АФК и митохондриями путем активации антиоксидантной системы, снижающей уровни АФК (рис. 4). Ядерный фактор эритроид-2-связанного фактора (NRF2) — ключевой регулятор транскрипции антиоксидантов, включая тиоредоксин-зависимую пероксидредуктазу (PRDX3), которая нейтрализует большую часть образующейся в митохондрии перекиси водорода [114,115]. Активность NRF2 снижается с возрастом [116]. Прогерин же связывает NRF2 и тем самым ингибирует его транскрипционную активность, что приводит к хроническому оксидативному стрессу и старению клетки при СПХГ [27]. Дефекты антиоксидантных защитных механизмов лежат в основе болезней Альцгеймера и Паркинсона, что, вероятно, обусловлено либо нарушениями активации NRF2, либо же непосредственным ингибированием антиоксидантов патологическими белками — к примеру, ингибирование PRDX3 мутантным белком LRRK2, приводящим к болезни Паркинсона [108,115,117]. Восстановление митохондриальной целостности посредством оверэкспрессии каталазы — митохондрия-адрессованного антиоксиданта — увеличивает продолжительность жизни мышам и оказывает протективное действие в отношение нейродегенерации, болезней сердца, онкологических заболеваний и инсулинорезистентности [103,118].
Усиленное обновление митохондрий и замещение поврежденных митохондрий интактными также защищает от оксидативного повреждения. Коактиватор-1α γ-рецептора, активируемого пролифераторами пероксисом (PGC1α) способствует митохондриальному биогенезу, а параллельно этому активирует NRF2-опосредованный антиоксидантный ответ (рис. 4) [103]. Активация PGC1α уменьшает тяжесть митохондриальной дисфункции и других дефектов старения в фибробластах пациентов с СПХГ [105], несмотря на то, что, судя по всему, положительные эффекты активации PGС1α in vivo являются тканеспецифичными и зависят от длительности и степени активации [37]. Сниженная активность PGС1α наблюдается при болезнях Альцгеймера и Паркинсона и сопричастна развитию связанной с ХБП мышечной дистрофии [110,119]. Наблюдаемое при СПХГ, старении и AADs снижение активности PGC1α вероятнее всего и объясняется недостаточной активацией SIRT1 и АМФК (вышестоящих регуляторов PGС1α) [26].
Таким образом, эти наблюдения показывают, что метаболический сигналинг связывает между собой митохондриальную целостность и образование АФК. Баланс в этой триаде оказывает большое влияние на протеолитическую систему аутофагии, участвующую в деградации дефектных митохондрий, обороте аминокислот, требуемых для регулируемого mTOR синтеза белков, и элиминации поврежденных АФК макромолекул в целях поддержания гомеостаза клетки.
Протеостаз и протеолиз
Под протеостазом понимается поддержание функционального протеома путем сбалансированной регуляции синтеза белка, восстановления его структуры и протеолиза. Целостность протеома зависит от беспрерывного обновления белков, заключающегося в деградации поврежденных и неверно уложенных пептидов посредством аутофагии и убиквитин-протеосомной системы (УПС), а при прогероидных синдромах человека, физиологическом старении и AADs нарушается и то, и другое.
Убиквитин-протеасомная система (УПС). Комплекс, обеспечивающий АТФ-зависимую деградацией белков, помеченных специальной меткой — убиквитином.
Аутофагия уменьшает связанные со старением дефекты при СПХГ
Метаболически обусловленное накопление поврежденных митохондрий и агрегатов окисленных белков при старении и при патологиях говорит о дисбалансе между действием протеотоксических факторов и протеолитических путей, уничтожающими поврежденные органеллы и белковые агрегаты. Аутофагия является главным механизмом деградации, при котором аутофагосомы поглощают поврежденные компоненты клетки и способствуют их деградации посредством слияния с лизосомами, содержащими гидралазу [120].
Любопытно, что каскад IIS ингибирует аутофагию путем активации mTOR (рис. 4) [26]. Тогда как временная приостановка аутофагии, вызванная mTOR, может быть полезна для здоровья клетки, способствуя анаболизму, длительное ингибирование аутофагии приводит к накоплению токсичных белковых агрегатов в тельцах включения, т. н. агресомах, устойчивых к деградации. Агресомы содержат адапторный белок аутофагии p62 (известный также как SQSTM1) и убиквитин, которые в норме притягивают белковые агрегаты и способствуют их протеолизу [120]. При СПХГ были обнаружены убиквитин- и p62-положительные агрегаты прогерина [121]. Считается, что прогериновые агрегаты вредны для клетки из-за способности включать в свой состав нормальные клеточные белки, в том числе и NRF2 [27]. Активация аутофагии путем рапамицин-опосредованного ингибирования mTOR способствует повышению растворимости и деградации прогерина, тем самым уменьшая тяжесть дефектов при клеточном старении [121]. Терапия рапамицином увеличивает продолжительность жизни как у мышей, так и у C. elegans; причем этот эффект, вызванный ингибированием IIS, зависим от аутофагии [26,122].
Дефекты протеостаза и аутофагии при AADs
Существует доказательства положительного влияния стимуляции аутофагии при AADs, и эти эффекты большей частью можно свести к повышению утилизации патогенных токсических агрегатов, образующихся под действием хронического оксидативного стресса [120]. При болезни Альцгеймера уровень лизосомальной протеазы, катепсина D, снижен, из-за чего нарушается деградация β-амилоида и агрегатов фосфорилированного тау-белка. Ослабление гиперактивированного сигналинга IIS-mTOR повышает интенсивность аутофагии этих агрегатов и уменьшает выраженность нарушений памяти у мышиных моделей болезни Альцгеймера [123,124]. У лиц с болезнью Паркинсона снижены уровни лизосом-ассоциированного мембранного гликопротеина 2 (LAMP2), который, предположительно, препятствует протеолизу α-синуклеиновых агрегатов и способствует смерти нейронов [125]. Вдобавок к этому, генетические дефекты в белках parkin и PTEN-индуцированной киназы 1 (PINK1) (см. Вставка 1) снижают активность аутофагии поврежденных митохондрий при семейной болезни Паркинсона [120].
Дефицит LAMP2A и катепсина D является также известной причиной кардиомиопатий, а ослабление аутофагии может способствовать атеросклерозу вследствие активации клеточной смерти и старения ГМС [126,127]. Агрегация белков также участвует и в патогенезе СД2: было обнаружено, что в поджелудочной железе у мышиных моделей СД2 образуются агрегаты амилина; более того, агрегация усиливается при генетической абляции белка аутофагии ATG7 [128]. Кроме этого мутация, повышающая способность амилина образовывать агрегаты, также ассоциирована с СД2. Агрегация мутантного амилина предшествует развитию дисфункции панкреатических β-клеток и этого достаточно, чтобы индуцировать диабетический фенотип у крыс дикого типа [128].
Низкая протеосомная активность при старении и AADs
УПС обеспечивает деградацию поврежденных белков путем АТФ-зависимого протеолиза [126]. У пациентов с СПХГ ее активность снижена (рис. 4) [129], и она так же снижается в ходе старения. С этим согласуются полученные данные о том, что повышенная активность УПС связана с увеличением продолжительности жизни Drosophila melanogaster и C. elegans [129].
Дофаминэргическая абляция субъединицы P26S4 УПС у мышей приводит к усилению агрегации α-синуклеина и к гибели нейронов, подобно тому, как это происходит при болезни Паркинсона [120]. Вдобавок к этому, пониженные уровни PA700 и PA28, активаторов УПС, в черном веществе коррелируют со степенью агрегации α-синуклеина у пациентов с болезнью Паркинсона [130]. Умеренное снижение активности УПС путем оверэкспрессии мутантной доминантно-негативной каталитической субъединицы протеасом PSMB5 усугубляет поражения сердца, вызванные ишемией-реперфузией, а повышение уровня пре-амилоидных олигомеров сопровождает дилатационную и гипертрофическую кардиомиопатии [126]. Соответственно, стимуляция УПС — перспективная терапевтическая стратегия для лечения кардиомиопатий [126]. Таким образом, данные наблюдения говорят нам о том, что аутофагия и УПС играют ключевую роль в противодействии связанному со старением метаболически индуцированному стрессу.
Сдвиги в судьбе клетки
Аккумуляция клеточных повреждений при старении приводит к запуску сигнальных путей, контролирующих пролиферацию и дифференцировку клеток, равно как и опосредованную стволовыми клетками регенерацию, в целях предупредить постоянный коллапс гомеостаза на клеточном и тканевом уровнях. Особенно важное место в старении и развитии AADs занимает клеточное старение и пути регенерации.
Усиление клеточного старения при СПХГ и AADs
Потеря геномной и протеомной целостности может запускать клеточное старение, являющееся, по сути, необратимым арестом пролиферации, главным образом зависимым от путей супрессии опухоли RB-p16 и p53-p21 (рис. 5) [131,132]. Временная активация этих путей может иметь позитивное значение, поскольку она позволяет восстановить клеточные повреждения прежде, чем продолжится пролиферация. Тем не менее, длительная активация RB-p16 и p53-p21 приводит к негативным последствиям, потому что она запускает самоподдерживающийся воспалительный сигналинг, обусловленный трансформирующим фактором роста-β (ТФР-β) и ядерным фактором каппа-B (NF-κB). Этот сигналинг характеризуется экспрессией интерлейкина-1 (ИЛ-1) и ИЛ-6, фактора некроза опухоли-α (ФНО-α), циклооксигеназы 2 (ЦОГ-2) и хемокином с мотивом C-X-C 1 (CXCL1) и CXCL2 (рис. 5) [131]. Это состояние получило название связанный со старением секреторный фенотип (SASP), и долговременная активация данного сигналинга может нарушить коммуникацию между стареющими и иммунными клетками, тем самым предотвращая опосредованную иммунной системой элиминацию стареющих клеток, которая в норме и запускается SASP [131]. Невозможность уничтожения стареющих клеток может еще больше усугубить возрастное нарушение физиологических функций, индуцируя клеточное старение в соседних клетках посредством щелевых контактов [26]. Не противоречит этому наблюдение, что искусственно вызванная элиминация стареющих клеток увеличивает продолжительность и качество жизни прогероидных мышей с отсутствием белка контрольной точки митоза BubR1 (табл. 1), препятствует атерогенезу у мышей с дефектом рецептора к липопротеинам низкой плотности и оказывает протективное действие на почки и сердце у мышей дикого типа [6,7,133].
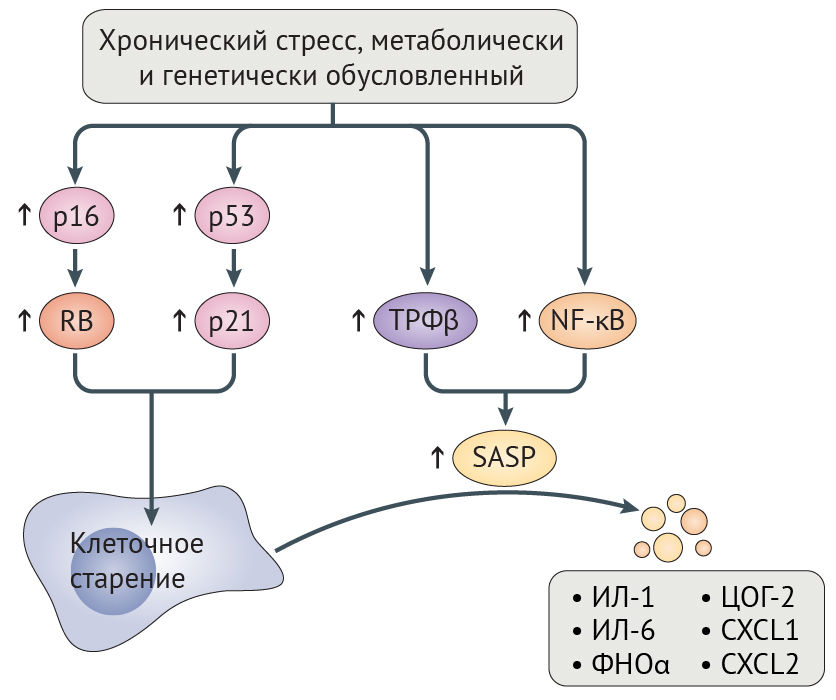
ЦОГ-2 — циклооксигеназа 2; CXCL1 — хемокин с мотивом C-X-C 1; ФНО-α — фактор некроза опухоли-α.
При СПХГ увеличенное количество клеточных повреждений приводит к гиперактивации RB-p16, p53-p21 и NF-κB, своим следствием имея клеточное старение и SASP, что сопровождается повышением уровней ИЛ-6, ФНО-α, CXCL1 и CXCL2 у мышиных моделей прогерии [34,134,135]. Гиперактивация NF-κB отчасти вредна из-за эпигенетических эффектов, поскольку это активирует гистон-метилтрансферазу DOT1L [136]. И наоборот, инактивация NF-κB или p53 на генетическом уровне увеличивает продолжительность и качество жизни у мышиных моделей СПХГ [34,134].
При болезни Альцгеймера поддерживающие нейроны астроциты, которые обладают естественной высокой базальной активностью NF-κB и вовлечены в патогенез данного заболевания, демонстрируют повышенную экспрессию p16 и ИЛ-6 [137]. Гиперактивация NF-κB по каноническому пути пагубна для астроцитов и, судя по всему, способствует агрегации β-амилоида, в свою очередь обратимо запускающего опосредованные ФНО-α, ИЛ-1β и ЦОГ-2 воспалительные сигнальные каскады [138]. Болезнь Паркинсона, аналогично болезни Альцгеймера, ведет к повышению активности NF-κB и уровней ЦОГ-2 [139]. Терапия мышей дикого типа ингибиторами ЦОГ-2 нивелирует нейротоксический эффект МФТП (1-метил-4-фенил-1,2,3,6-тетрагидропиридина), инициирующего болезнь Паркинсона посредством ингибирования OXPHOS [139]. Повышенные уровни ИЛ-1β далее усугубляют прогрессирование болезни Паркинсона, индуцируя экспрессию α‑синуклеина [139].
При старческом идиопатическом фиброзе легких повышение содержания ИЛ-1β может активировать дифференцировку фибробластов легких в миофибробласты, тем самым поддерживая фиброз [140]. SASP-регулируемые изменения клеточных характеристик играют роль и в злокачественных процессах, в силу того, что ИЛ-6 и ИЛ-8 вносят вклад в развитие опухолей, содействуя эпителиально-мезенхимальному переходу (ЭМП) [131]. Ингибиторы ТФР-β, являющиеся супрессорами SASP, продемонстрировали способность подавлять прогрессию некоторых опухолей и в настоящее время проходят клинические испытания [141]. Наконец, ТФР-β- и NF-κB-обусловленные воспалительные ответы, ведущие к SASP, участвуют в патогенезе ХБП, и репрессия обоих путей ингибиторами ангиотензин-превращающего фермента замедляет развитие ХБП [141].
Эпителиально-мезенхимальный переход (ЭМП). Процесс, в ходе которого эпителиоциты претерпевают различные изменения на молекулярном уровне: изменяется характер межклеточных контактов, полярности клетки и способность к инвазии, с целью превращения в мезенхимальные клетки. ЭМП играет положительную роль при заживлении ран, но в то же время и негативную — при фиброзе внутренних органов и инициации метастазирования опухолей.
В целом, необратимая остановка роста и устойчивая гиперактивация воспалительных секреторных сигнальных путей ведут к перманентному состоянию клеточного старения, лежащего в основе старения и AADs. Накоплению стареющих клеток может препятствовать регенерация посредством стволовых клеток.
Нарушение регенеративной способности стволовых клеток
Стволовые клетки, которые способны к самообновлению и мультилинейной дифференцировке, располагаются в специфических нишах по всему человеческому телу. Человеческие мезенхимальные стволовые клетки (МСК), присутствующие фактически во всех тканях, дифференцируются в остеоциты, хондроциты и адипоциты, в то время как ГМС, не окончательно дифференцированные клетки сосудистой стенки, обладают ограниченным количеством путей дифференцировки [142,143]. Связанные со старением дефекты фибробластов, взятых от пациентов с СПХГ, могут быть обращены вспять путем преобразования фибробластов в индуцированные плюрипотентные стволовые клетки (иПСК), не экспрессирующие прогерин из-за естественной репрессии транскрипции гена LMNA [30, 31]. После повторной дифференцировки СПХГ-иПСК в МСК и ГМС реактивация транскрипции гена LMNA и, соответственно, индукция экспрессии прогерина ведет к появлению типичных геномных, протеомных и клеточных дефектов старения, характерных для СПХГ (рис. 1–4) [30,31], и снижает выживаемость при таких стрессовых состояниях как гипоксия. Эта зависимость может объяснить потерю ГМС у пациентов с СПХГ при гемодинамическом стрессе, равно как и сниженную способность СПХГ-МСК предотвращать повреждения в ходе ишемии-реперфузии на мышечной модели задней конечности [31]. Выживаемость и способность СПХГ-МСК противостоять хроническому оксидативному стрессу может быть восполнена посредством повторной активации NRF2, ингибируемого прогерином [27].
В дополнение к истощению пула стволовых клеток, которое также может быть характерно для стволовых клеток дермы у мышиной модели СПХГ, накопление дефектов старения может нарушать регенеративную способность стволовых клеток путем изменения их потенциала дифференцировки [144]. Так прогерин задерживает дифференцировку ГМС и способствует остеогенной дифференцировке МСК за счет усиленного образования адипоцитов, что может способствовать кальцификации тканей (таких как кожа и стенки сосудов), наблюдаемой у пациентов с СПХГ [48,145].
Наблюдаемое у стареющих пациентов накопление небольших количеств прогерина и малое содержание белка Вернера в ГМС и МСК дикого типа, соответственно, также может объяснить снижение регенеративной способности стволовых клеток в ходе физиологического старения [82,146]. Действительно, сообщается, что в ходе старения в МСК, ГМС, гемопоэтических стволовых клетках (ГСК), клетках скелетных мышц и нервных стволовых клетках наблюдаются один или более типичных признаков клеточного старения, таких как оксидативный стресс или активация сигнальных путей повреждения ДНК, старения и SASP [143,147]. Негативные эффекты этих признаков на стволовые клетки наглядно проявляются ухудшением восстановления тканей легких и поджелудочной железы, в норме обладающих высоким регенеративным потенциалов, у пожилых людей после частичной резекции — по сравнению с молодыми пациентами [148, 149]. Таким образом, сайленсинг p16, стимуляция SIRT1, блокирование эффектов SASP путем оверэкспрессии антагониста рецептора ИЛ-1 или же уменьшение образование АФК путем оверэкспрессии антиоксиданта, супероксиддисмутазы 2, повышают регенеративную способность ГМС и ГСК, равно как и нервных, кишечных и мышечных стволовых клеток [143,147].
Взрослые стволовые клетки могут быть более подвержены возрастным дефектам, т.к. они в основном находятся в состоянии покоя (т. е. в состоянии временного ареста пролиферации) чему, по некоторым данным, сопутствуют снижение уровня белков репарации ДНК и отсрочка репарации ДНК до момента вступления в новый клеточный цикл [147]. По всей видимости, накопление повреждений ДНК во время этого периода покоя может пересечь ту пороговую черту, после которой начинается снижение функции стволовой клетки.
Снижение регенеративной способности стволовых клеток играет важную роль при некоторых AADs. К примеру, старение ГМС ведет к образованию атеросклеротических бляшек [150]. При СД2 МСК могут обладать сниженной способностью к пролиферации и дифференцировке, а активность МСК и панкреатоспецифических стволовых клеток играет важную роль в поддержании количества и функции β-клеток поджелудочной железы [148]. Более того, потенциал дифференцировки стволовых клеток легких нарушается при старческом идиопатическом легочном фиброзе, а функциональные МСК способствуют заживлению остеопоротических переломов [143,149]. Хотя и неизвестно, участвуют ли сердечные и нервные стволовые клетки в обновлении окончательно дифференцированных миоцитов и нейронов [147,151], их позитивные секреторные иммунорегуляторные эффекты установлены, а на мышиных моделях болезни Альцгеймера было продемонстрировано, что после инъекции МСК смягчаются когнитивные нарушения [147,151].
Таким образом, на основе этих данных о болезнях преждевременного старения, физиологическом старении и AADs можно предположить, что преждевременное старение и AADs обусловлены генетическими, метаболическими дефектами и дефектами клеточного старения, которые вызывают сдвиги в судьбе стволовых клеток.
Перспективы
В данной работе авторы проанализировали связь между преждевременным старением, нормальным старением и AADs. Обсудили, как каждый конкретный дефект в регуляции геномной, теломерной, эпигенетической, метаболической, митохондриальной и протеолитической целостности, а также целостности клеточного цикла, влияет на процесс развития клеточного старения и AADs (рис. 1). Представленный рисунок отображает одну из общих схем связи между этими ключевыми признаками старения; как можно видеть, нарушение одного из этих процессов вызывает появление дефектов в других, что в конечном счете ведет к коллапсу клеточного гомеостаза, обуславливающему болезнь. Хотя этот каскад уменьшает продолжительность здоровой жизни и запускает старение, тесное переплетение этих событий может предоставить возможностью лечения: терапии, направленной на один (или более) из этих признаков, может оказаться достаточно для облегчения процесса в целом.
Наблюдения, полученные в исследованиях систем преждевременного старения, нормального старения и AADs дополняют друг друга. Хотя прогероидные синдромы отражают только некоторые аспекты процесса старения, тем не менее, они представляют немалый интерес по причине четко детерминированных генетических признаков и возможности проведения контролируемых экспериментов.Физиологическое старение и AADs отражают действительный процесс старения, но их изучение сопряжено с трудностями интерпретации в контексте физиологии и среды. Чтобы дальше распутать эту, богатую тонкостями, сеть клеточного старения, потребуется разработка новейших экспериментальных модельных систем, которым не будет помехой традиционно медленное стохастическое накопление и низкая пенетрантность дефектов старения. Прогероидные синдромы у человека предоставляют такую возможность в силу того, что связанные дефекты клеточного старения во многом повторяют те, что наблюдаются при физиологическом старении и AADs. Более того, ускоренная природа патогенеза болезней преждевременного старения, их четко определяемая генетическая причина и их мощнейшая фенотипическая пенетрантность позволяют разработать надежные методы изучения клеточного старения.
Создание иПСК, полученных от пациентов с этими и другими болезнями преждевременного старения — новая многообещающая стратегия, дающая возможность в реальном времени наблюдать формирование уже известных возрастных дефектов [152]. В качестве альтернативы, индукция экспрессии мутантных белков, связанных с прогероидными синдромами в клетках дикого типа, может тоже ответить на эти вопросы, а обе стратегии хороши еще и тем, что их легко можно комбинировать с доступными высокопроизводительными скрининговыми методами РНК-интерференции (RNAi) и CRISPR. Использование системы редактирования генома CRISPR-Cas9 может ускорить описание недавно идентифицированных механизмов старения, способствуя созданию in vivo новых моделей старения и моделей AADs.
Авторы ожидают, что разработка новых экспериментальных методик для изучения болезней преждевременного старения человека начнет отвечать на ключевые вопросы о том, каким образом дефекты клеточного старения связаны между собой и как они взаимодействуют с путями, противодействующими старению. Подобные исследования улучшат понимание процесса старения и облегчат диагностику и лечение ведущих хронических заболеваний, связанных со старением, таких как болезни Альцгеймера и Паркинсона, атеросклероз, диабет и рак.
