
Ферроптоз

Ферроптоз — это железозависимая форма регулируемой гибели клеток, которая происходит в результате исчезновения активности липид-восстанавливающего фермента глутатионпероксидазы 4 (GPX4), окисления Fe (II) до Fe (III) и последующего накопления активных форм кислорода и липидных гидропероксидов, которые способны окислить практически любое вещество клетки.
Морфологически ферроптоз характеризуется сжатием клетки и наличием меньших по размеру митохондрий с увеличенной плотностью внутренней мембраны, уменьшением или полным исчезновением крист и разрывами внешней митохондриальной мембраны. Ключевыми соединениями, индуцирующими ферроптоз в опухолевых клетках и некоторых нормальных клетках (например, клетках почечных канальцев, нейронах, фибробластах и Т-клетках), являются блокаторы глутатионпероксидазы и вещества, снижающие содержание глутатиона в клетке. Глутатионпероксидаза катализирует восстановление липидных гидропероксидов в спирты и перекиси водорода в воду.
К индукторам ферроптоза можно отнести экспериментальные соединения (например, эрастин, RAS related lethal 3, RAS related lethal 5 и бутионинсульфоксимин) и клинические препараты (например, сульфасалазин, сорафениб и артесунат). Активация митохондриальных потенциал-зависимых анионных каналов и митоген-активированных протеинкиназ, усиление стресса эндоплазматического ретикулума и ингибирование антипорта глутамат/цистин участвуют в индукции ферроптоза. Этот процесс характеризуется накоплением продуктов перекисного окисления липидов и активных форм кислорода (АФК), полученных из окисления железа в результате реакции Фентона (рис. 1), и может быть фармакологически ингибирован хелаторами железа (например, дефероксамином и десфериоксамином мезилата) и ингибиторами перекисного окисления липидов (например, ферростатином, липоксстатином и зилеутоном).
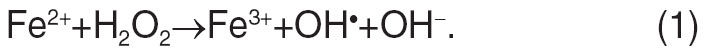
Рисунок 1 | Реакция Фентона.
Глутатионпероксидаза — 4, белок теплового шока бета-1 и ядерный фактор (NFE2L2) функционируют как отрицательные регуляторы ферроптоза, ограничивая производство АФК и снижая поглощение железа клетками. Оксидаза NADPH и p53 (особенно мутация с дефектами ацетилирования p53) действуют наоборот: как положительные регуляторы ферроптоза путем увеличения продукции АФК и ингибирования экспрессии SLC7A11 (специфической легкой цепи субъединицы антипорта глутамат/цистин). Неконтролируемый ферроптоз связан с несколькими физиологическими и патологическими процессами, включая гибель опухолевых клеток, нейротоксичность, нейродегенеративные заболевания, острую почечную недостаточность, гепатотоксичность, вызванную лекарственными средствами, повреждение печени и сердца в результате ишемии/реперфузии, а также нарушением Т-клеточного иммунитета.
Механизмы ферроптоза, ключевые регуляторы и мишени их действия
Эрастин и сульфасалазин блокируют антипорт глутамат/цистин. Количество цистина, который является предшественником синтеза глутатиона, в клетке снижается, соответственно снижается количество глутатиона — субстрата фермента GPX4, который окисляется до дисульфида глутатиона и восстанавливает перекись водорода до воды. Бутионинсульфоксимин (BSO) снижает количество глутамина, а также ингибирует фермент глутамат-цистеин лигазу, лимитирующий скорость синтеза глутатиона. Снижение количества глутатиона приводит к снижению антиоксидативной способности клетки и накоплению АФК. Ферроптоз, индуцированный RSL3, имеет общие черты с ферроптозом, вызванным эрастином. Однако RSL3 не ингибирует антипорт глутамат/цистин и не уменьшает количество глутатиона. Вместо этого RSL3 ингибирует главный фермент — GPX4. Несмотря на разные мишени, RSL3 и эрастин вызывают ферроптоз по похожим механизмам. Сорафениб является мультикиназным ингибитором и используется для лечения карциномы почки и гепатоцеллюлярной карциномы. В клетках последней доказано, что сорафениб активирует ферроптоз. Сорафениб, также как эрастин, блокирует антипорт глутамат/цистин, однако выяснилось, что в ответ на сорафениб повышается содержание белка ретинобластомы, который подавляет ферроптоз. FIN56 (от ferroptosis-inducing agents — вещества, индуцирующие ферроптоз) и CIL56 являются небольшими молекулами, которые вызывают повышение количества АФК путем посттрансляционной деградации GPX4, а также блокируя синтез липофильных антиоксидантов, таких как коэнзим Q10.
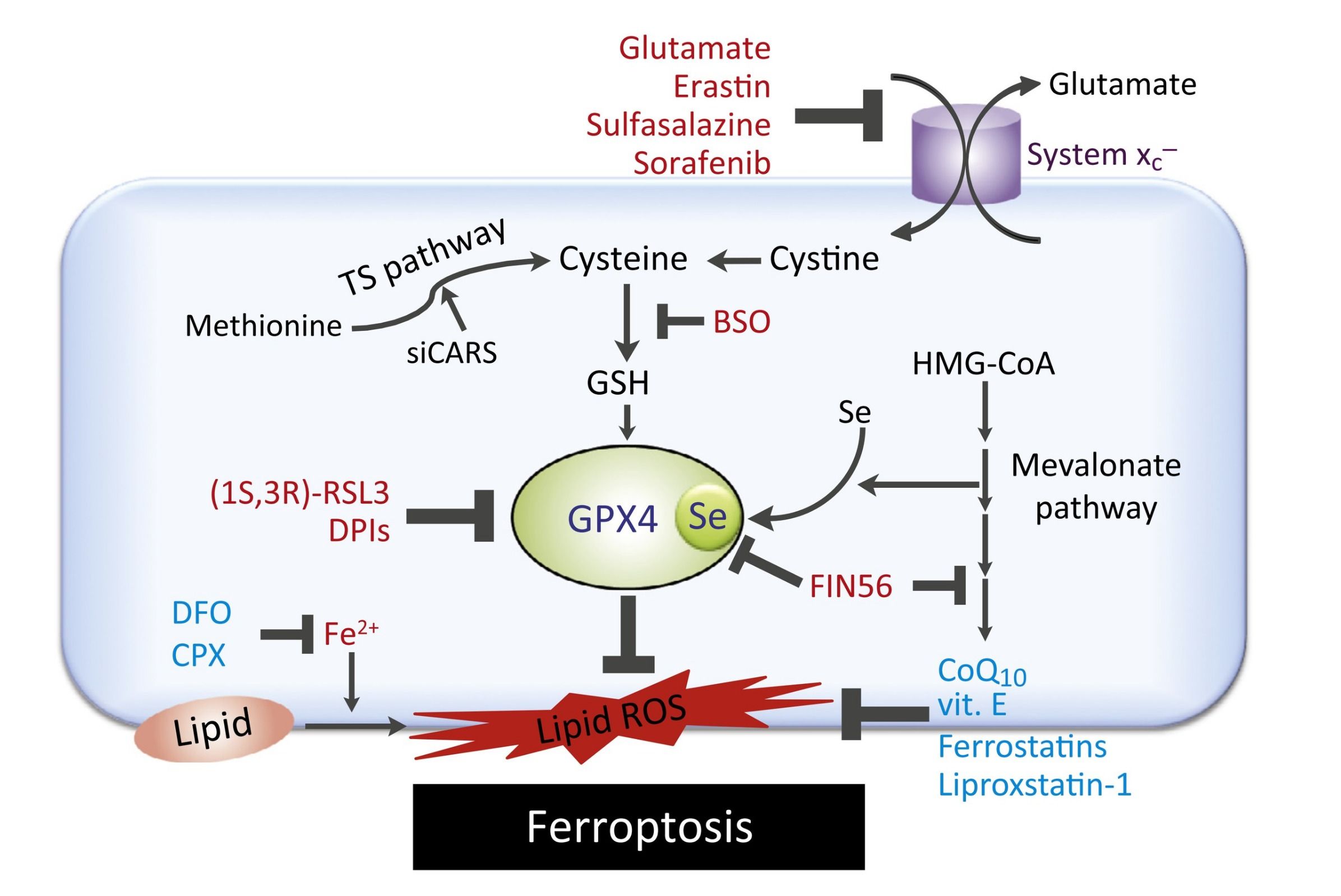
Схема 1 | Индукторы ферроптоза обозначены красным цветом, ингибиторы - синим. TS pathway - транссульфирование, Se - селеноцистеин, DFO - дефероксамин, CPX - циклопироксоламин, CoQ10 - коэнзим Q10.
Пути регуляции ферроптоза
Мевалонатный путь
GPX4 является селенопротеином (содержит селеноцистеин). Биосинтез селенопротеинов регулируется мевалонатным путем. Ингибиторы мевалонатного пути, такие как статины, взаимодействуют с тРНК селеноцистеина, что влияет на созревание и биосинтез GPX4. Попытки влияния на мевалонатный путь при гиперхолестеринемии, остеопорозе, онкологии показывают, что последующие исследования в регуляции ферроптоза пойдут по этому пути.
Транссульфирование
Цистеин — заменимая аминокислота, поскольку может образовываться путем транссульфирования, донором серы является метионин. Помимо синтеза de novo, клетки, используя антипорт глутамат/цистин, получают предшественник цистеина, восстанавливая количество аминокислоты необходимой для синтеза белков и обезвреживания АФК. Метаболиты транссульфирования, такие как цистеин и цистатионин, активируют цистеинил тРНК синтетазу (cysteinyl tRNA syntetase — CARS), которая является генетическим супрессором ферроптоза, вызванного эрастином. Транссульфирование становится жизненно необходимым для клетки, когда ингибируются механизмы поглощения цистина при влиянии эрастина. Ингибирование этого пути вместе с влиянием эрастина было бы летальным для опухолевых клеток.
Дополнительные пути регуляции
Глутамин и трансферрин могут индуцировать ферроптоз при сывороточной депривации путем глутаминолиза. Более того, ингибирование глутаминолиза носило защитный характер при повреждении сердца в результате ишемии/реперфузии, что предполагает, что это еще одна дополнительная стратегия для подавления ферроптоза. Белок теплового шока бета-1 подавляет ферроптоз, что делает его мишенью для регулирования чувствительности.
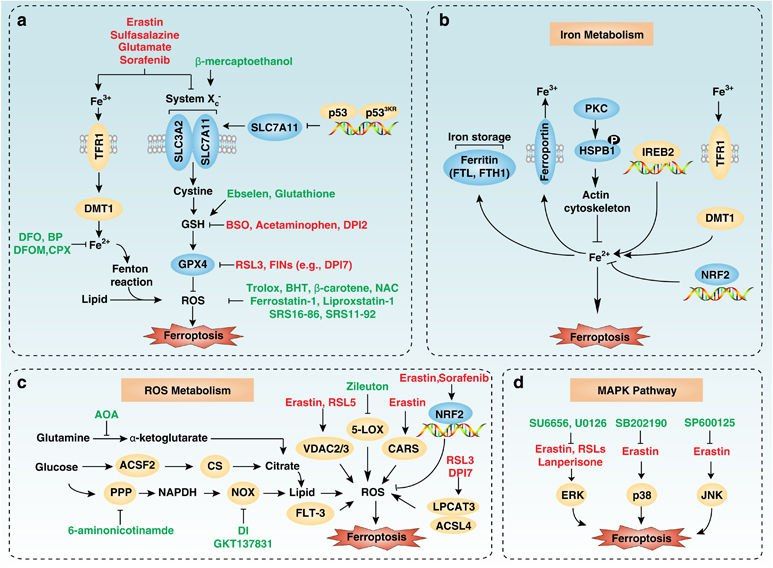
Схема 2 | Молекулярные механизмы и сигнальные пути ферроптоза.
Роль ферроптоза в развитии
Исследования на мышах показали, что полное отсутствие фермента GPX4 ведет к летальному исходу. Некоторые ткани и клетки, преимущественно нейроны головного мозга, накапливали липидные гидропероксиды и АФК, на которые может повлиять введение антиоксиданта витамина Е. Избирательное удаление GPX4 в нейронах привело к избирательной и моментальной дегенерации моторных нейронов по типу ферроптоза, наступил паралич. Предполагается, что ферроптоз может вызывать некоторые нарушения двигательных нейронов. Клетки почечных канальцев также чувствительны к индукции ферроптоза. Исключение GPX4 в мышиных Т-клетках вызвало ферроптоз, что привело к недостаточному иммунному ответу на инфекцию.
Связь с заболеваниями человека
Наследственная спондилометафизарная дисплазия — аутосомно-рецессивное заболевание, характеризующееся нарушением роста костей и летальностью. Секвенирование генома ребенка и его семьи показывает мутации GPX4, которые ведут к недостатку активности фермента. Вследствие рабдомиолиза высвобождаются миоглобин и железо гема, что приводит к образованию АФК и острому повреждению почек. Использование ферростатина-1, ингибитора ферроптоза, предотвратило смерть клеток в проксимальных почечных канальцах, что указывает на развитие ферроптоза в них при рабдомиолизе. Такие же результаты были показаны при остром повреждении почек, вызванном кристаллами оксалата. Ферростатин третьего поколения SRS16-86 при повреждении, вызванном ишемией/реперфузией, смог оказать защитное действие на функцию почки и дальнейшее выживание мыши. Специфический ингибитор ферроптоза liproxstatin-1 подавляет ферроптоз, вызванный эрастином, RSL3, BSO, а также увеличивает выживаемость мышей с острым повреждением почек. Эти результаты указывают на восприимчивость ткани почек к ферроптозу и предполагают эффективность использования его ингибиторов.
Хорея Гентингтона — генетическое заболевание нервной системы, развивающееся при увеличении повторов триплетов ЦАГ (цитозин-аденин-гуанин) более 36 в гене HTT, который кодирует белок хантингтина (Htt). Если триплетов становится больше 36, то синтезируется удлинённый полиглутаминовый тракт и происходит образование мутантного белка хантингтина (mHtt), оказывающий токсичное действие на клетки. От количества повторов зависит пенетрантность болезни. Токсическое действие связано с нарушением глутатионовой регуляции окислительно-восстановительных процессов и метаболизма внутриклеточного железа. Внутрижелудочковое введение дефероксамина, хелатора железа, у мышиных моделей болезни, показало протективный эффект нейронов. Ферростатин-1 предупреждал смерть нейронов, вызванную геном HTT с патологическим повтором. Эти данные поддерживают гипотезу, что ферроптоз повреждает клетки при болезни Гентингтона.
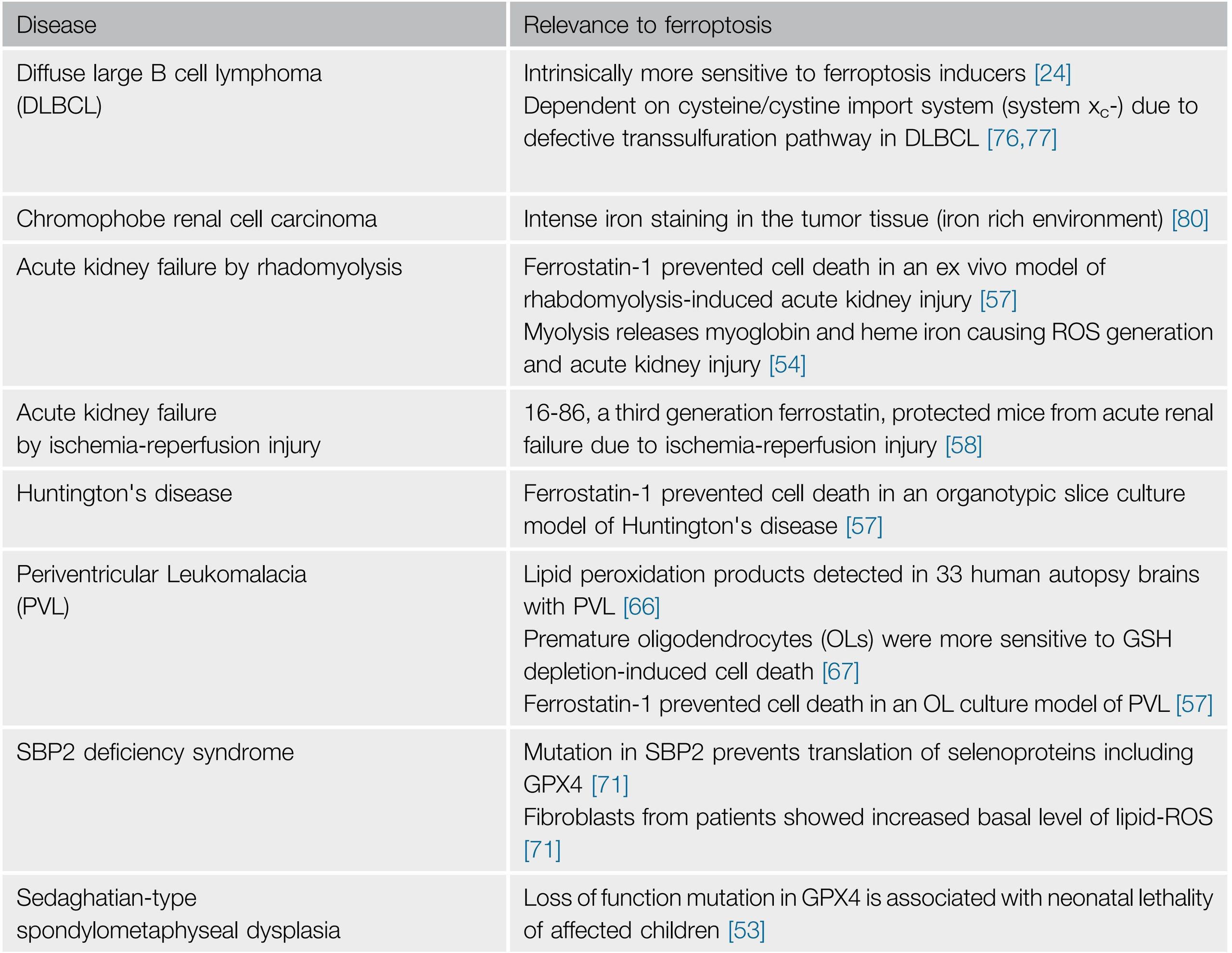
Таблица 1 | Ферроптоз при патологических состояниях.
Перивентрикулярная лейкомаляция (ПВЛ) — форма поражения белого вещества полушарий головного мозга у детей, открытая морфологами, одна из причин детского церебрального паралича. На клеточном уровне это связано с потерей олигодедроцитов. Анализ образцов цереброспинальной жидкости пациентов показал наличие большого количества продуктов окисления липидов, таких как 8-изопростан и малондиальдегид. На мышиных моделях было обнаружение снижение количества глутатиона, приводящее к клеточной смерти. Однако, витамин Е, ферростатин-1 и его аналоги показали протективное действие на олигодендроциты, что предполагает ферроптоз в данном случае.
Большие дозы парацетамола индуцировали ферроптоз в гепатоцитах, снижая количество глутатиона, что, предположительно, является механизмом токсического воздействия лекарств (как минимум этого препарата). Феростатин-1 и его аналоги могут ингибировать гибель клеток, вызванную парацетамолом. Liproxstatin-1, упоминавшийся выше, также улучшает состояние клеток печени при их повреждении, вызванном ишемией/реперфузией.
Диффузная B-крупноклеточная лимфома и карцинома почки крайне подвержены к ферроптозу, индуцированному эрастином и сорафенибом, поскольку имеют дефекты транссульфирования. Улучшенные аналоги эрастина, такие как piperazine erastin и imidazole ketone erastin, могут использоваться как пробы опухолевой чувствительности к ферроптозу. Артемизинин индуцирует клеточную смерть, связанную с железом, в том числе и ферроптоз, поэтому он, предположительно, будет эффективен при лечении опухолей, которые кумулируют железо. RSL3 и RSL5 в Ras-мутантных клетках оказываются неэффективны из-за активации NRF2, который увеличивает количество антиоксидантных белков и защищает опухолевые клетки от ферроптоза.
Недавно было идентифицировано несколько молекул для регулирования ферроптоза прямым или косвенным воздействием на метаболизм железа и перекисное окисление липидов. Эти так называемые регуляторы ферроптоза также участвуют в других типах регулируемой клеточной смерти. Таким образом, самая важная задача при изучении ферроптоза заключается в выявлении нисходящих сигнальных путей и ключевых ферментов железозависимого метаболизма АФК, чтобы отличить ферроптоз от других типов клеточной смерти. Дальнейшее определение генотипически-селективной активности ферроптоза при опухолях и задействованных механизмов будет важно для выбора терапии на основе ферроптоза. Он также играет важную роль при стерильных воспалительных процессах, таких как острая травма тканей, повреждение, вызванное ишемией/реперфузией и нейротоксичности. Более глубокое понимание роли ферроптоза при онкологических и других заболеваниях создаст новые возможности для диагностики и лечения.
Источники
-
Yang W. S., Stockwell B. R. Ferroptosis: death by lipid peroxidation // Trends in cell biology. — 2016. — Т. 26. — №. 3. — с. 165–176.
-
Xie Y. et al. Ferroptosis: process and function // Cell death and differentiation. — 2016. — Т. 23. — №. 3. — С. 369.
-
Вартанян А. А. Метаболизм железа, ферроптоз, рак // Российский биотерапевтический журнал. — 2017. — Т. 16. — №. 3.
