
Осколки спектра

Масштабные геномные исследования вновь привлекли внимание к небольшой группе расстройств аутистического спектра, которые диагностируются молекулярно-биологическими методами. Новые данные пробудили интерес к генетическим методам лечения РАС.
Этот год стал важной вехой для нескольких впервые проведенных на людях испытаний таргетных методов лечения расстройств аутистического спектра (РАС), включая генную терапию и применение антисмысловых олигонуклеотидов (АСО; см. таблицу 1). Следом ряд крупных биофармацевтических компаний, таких как Novartis, Roche, Biogen, Ionis, PTC Therapeutics, Sarepta Therapeutics и Amicus Therapeutics, анонсировали несколько программ по лечению РАС. В прошлом году Novartis объявила о сделке на сумму 75 млн долларов для получения прав на платформу регуляции генной экспрессии Sangamo Therapeutics, основанную на белках с цинковыми пальцами (ZFP). В перспективе сумма сделки может увеличиться до 720 млн долларов с учетом дальнейших этапов исследования. А в марте инвесторы вложили 40 миллионов долларов в новую компанию Jaguar Therapeutics (основана бывшими руководителями AveXis и занимается разработкой генной терапии), которая осуществляет реализацию программы лечения одной из генетически обусловленных причин аутизма: какой именно, пока не разглашается.
Таблица 1 | Генно-направленные препараты для лечения РАС в разработке

До относительно недавнего времени биология аутистических расстройств считалась недостаточно изученной, а само заболевание — слишком гетерогенным, чтобы разрабатывать для его лечения препараты молекулярной таргетной терапии. Теперь, в результате сотрудничества с Sangamo, Novartis оказывается в самом разгаре гонки за клинические разработки лечения РАС с помощью открытий в области геномики. На данный момент известно, что за развитие аутизма ответственны сотни генов. Накапливается уверенность в пенетрантности некоторых из них благодаря буму в сфере создания крупных геномных баз данных и появлению передовых лабораторных методов.
Однако долгий путь от исследований геномных связей до конкретных методов лечения только начинается. А с такими гетерогенными (как фенотипически, так и генетически) состояниями, как аутизм, на каждом этапе возникают трудности — от постановки правильного диагноза до поиска агентов, модифицирующих заболевание, которые смогут достигать пораженных участков мозга, и до определения биомаркеров результатов лечения и разработки клинических испытаний для их оценки.
На волне актуальности
Всемирная организация здравоохранения описывает аутизм как «ряд состояний, характеризующихся некоторой степенью нарушений коммуникации и речи, а также узким кругом интересов и занятий, которые являются уникальными для индивидуума и регулярно повторяются». По его оценкам, каждый 160-й ребенок в мире страдает РАС. Их распространенность растет [1], что побуждает некоторых исследователей говорить об «эпидемии». Однако остается неясным, насколько более точная диагностика и выявляемость приводят к увеличению числа случаев заболеваний.
Поиск генов, связанных с развитием РАС, ведется уже несколько десятилетий. Но большинство обнаруженных на сегодняшний день генов приводит к развитию заболевания лишь у небольшой части пациентов, а клинические проявления и степень выраженности симптомов очень разнообразны. У некоторых пациентов могут наблюдаться серьезные когнитивные нарушения или полное отсутствие речи. Другие кажутся практически здоровыми, пока не попадут в определенные обстоятельства. Как следствие, основной проблемой при разработке методов лечения РАС, основанных на геномных исследованиях, является сортировка всех данных о генах и фенотипах. Это необходимо для поиска мишеней, воздействие на которые приведет к наиболее выраженным фенотипическим изменениям.
К этой проблеме добавляется тот факт, что из-за нарушения развития нервной системы при РАС неясны сроки проявления заболевания, а также оптимальное время начала лечения. Даже изучение дебюта заболевания представляет сложность. По словам Стефана Сандерса, доцента психиатрии Института неврологии им. Вейля при Калифорнийском университете в Сан-Франциско, мозг в принципе трудно изучать, а для правильного понимания протекания аутизма необходимо исследование на этапах внутриутробного и постнатального развития.
Но есть два фактора, которые ускоряют поиск вариантов генов, склонных к мутациям. Первый — это доступ к постоянно растущим базам данных, посвященных непосредственно аутизму, в которых происходит сопоставление больших наборов признаков (см. таблицу 2). Второй — внедрение животных моделей и технологий для изучения процессов, лежащих в основе патологии, и поиска конкретных мишеней для терапии.
Таблица 2 | База данных по некоторым расстройствам аутистического спектра
- Autism Genome Project Consortium
50 исследовательских центров в Северной Америке и Европе; 4415 испытуемых - National Database for Autism Research
Источники лабораторных, клинических данных и данных исследований поведения, а также данных геномики и нейровизуализации - Autism Speaks MSSNG
Полногеномное секвенирование последовательностей ДНК 10 000 лиц, взятых из биобанка Autism Genetic Research Exchange - SFARI Gene
Данные из рецензируемых источников, разбитые на несколько блоков: блок генов человека (список генов со ссылками), блок вариантов генов по числу копий, животные модели, взаимодействие белков, анализ генов - AutDB Autism Informatics Portal
Гены человека, животные модели, взаимодействия белков и вариантов генов по числу копий, блоки анализа генов
Генетическая основа
Несмотря на то, что подозрения относительно генетической природы аутизма существовали давно, сегодня исследователи и клиницисты уже уверенно называют его генетическим заболеванием. Свен Сандин, эпидемиолог, специалист по этиологии аутизма и связанных с ним расстройств психического развития из Маунт Синай в Нью-Йорке, поясняет, что в 1970-х и 80-х годах клиницисты пришли к пониманию этого, основываясь преимущественно на небольших исследованиях близнецов. Ученый соглашается с тем, что это было хорошее начало. Но проблема с такими исследованиями заключалась в маленькой выборке испытуемых, а также большом количестве допущений от самих авторов, к примеру, о том, что близнецы росли вместе и получали одинаковое воспитание.
Сандин относительно недавно принимал участие в большом коллаборативном исследовании, участниками которого стали более чем два миллиона детей в возрасте от 0 до 15 лет из пяти стран — Дании, Финляндии, Швеции, Израиля и Австралии. Наблюдение за детьми осуществлялось в период с 1998 по 2012 год. За этот период РАС развились более чем у 22 тыс. из них [2]. Изучая подробные истории болезни этих детей, исследователи определили, что риск развития аутизма связан с генетикой почти на 80 %. Сандин поясняет, что в течение долгого времени среди этиологических факторов развития аутизма искали определенные факторы окружающей среды, такие как вакцинные препараты или воздействие на материнский организм таких факторов, как пестициды. Однако данное исследование стало крупнейшим в своем роде, и в ходе него удалось найти серьезные доказательства генетической природы аутизма.
Обратная генетика и мишени воздействия лекарственных средств
С РАС связано более тысячи генов [3], но исследователи сузили их число примерно до ста. Это те гены, которые, по-видимому, играют существенную роль в аутизме, а также в других расстройствах развития нервной системы. Крупнейшее на данный момент генетическое исследование по секвенированию генов аутизма — Autism Sequencing Consortium. Несколько научных групп консорциума выполнили полноэкзомное секвенирование генов более 35 000 человек, из них у почти 12 000 наблюдались РАС. Было определено 102 гена, увеличивающих риск развития РАС. Хотя некоторые из этих 102 генов были уже известны, 30 оказались новыми. Они не фигурировали в каких-либо исследованиях de novo или не являлись редкими вариантов других генов (рис. 1), [4]. Проведя анализ генной онтологии и данных литературы, группы выявили, что большинство этих генов задействованы в регуляции генной экспрессии, коммуникации и функционировании цитоскелета нейронов.
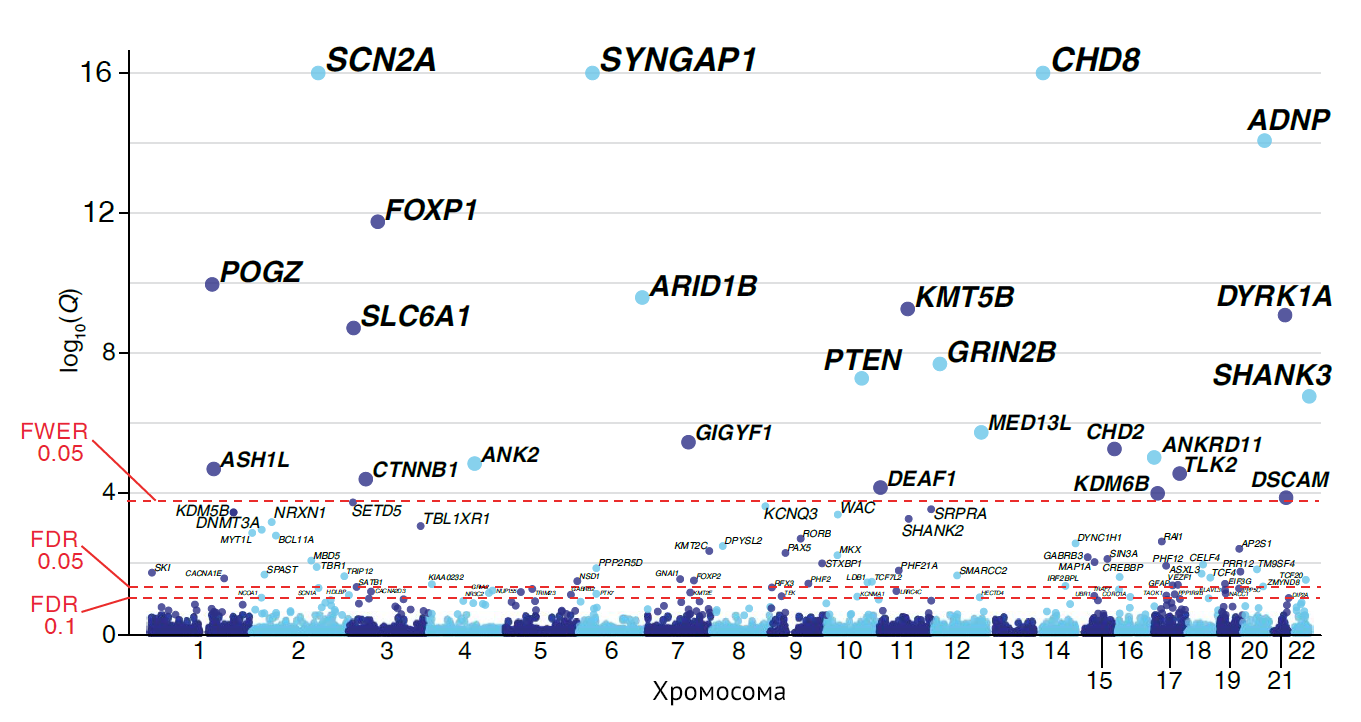
Сандерс, будучи членом консорциума, подробно изучал те гены, связь которых с развитием аутизма оказалась наиболее тесной. Этим генам соответствуют наименьшие значения p-value. На рис. 1 отрицательные логарифмы этих наименьших значений p-value — вершины точечной диаграммы. Такой тип диаграмм называется манхэттенским графиком: вершины — это небоскребы над массой более низких зданий — прим. ред. По мнению Сандерса, в этих генах должны быть более распространенными мутации с наибольшей пенетрантностью. Проведен функциональный анализ многочисленных миссенс-мутаций, обнаруженных у аутичных пациентов, в одном из подобных генов — SCN2A. SCN2A кодирует альфа-субъединицу 2 потенциал-зависимого натриевого канала. Группа Сандерса показала, что все варианты гена, найденные у аутичных детей, блокировали или снижали возбудимость нейронов. Они сделали вывод, что это может способствовать развитию аутизма [5]. Варианты этого же гена выявляются у пациентов с эпилепсией. Однако тот же функциональный анализ показал, что это приводит к противоположному эффекту — усилению возбудимости нейронов. Это демонстрирует важность функциональных исследований для понимания, приводит ли тот или иной вариант гена к потере или усилению функции. Предполагается, что при разработке методов лечения этих состояний [РАС и эпилепсии] требуется соблюдать осторожность, поскольку у значительной доли аутичных детей (20 %) возникают припадки.
CHD8 — другой ген, связанный с развитием РАС, стал предметом исследования ученых Вашингтонского университета в Сиэтле. Внимание на этом гене было сосредоточено, так как ранее удалось обнаружить связь между деструктивными мутациями гена и развитием РАС. Руководитель группы Эван Эйхлер описывает это исследование как «почти обратную генетику». В ходе исследования было проведено прицельное секвенирование гена CHD8 у порядка 6100 детей с аутизмом или фенотипами, характеризующимися задержкой развития. Это позволило выявить 15 независимых вариантов гена, не обнаруженных ни у 2289 их здоровых сиблингов, ни у 6503 детей из контрольной группы. Большинство детей с этими мутациями обладали схожей внешностью, у них наблюдались нарушения сна и желудочно-кишечные расстройства, которые часто встречаются у детей с РАС. Используя в качестве модели рыбку данио-рерио, ученые смогли изучить роль данного гена в развитии нервной системы. Повреждение гена CHD8 у рыб привело к увеличению головы, широко расставленным глазам, запорам и нарушениям пищеварения. Как обнаружили ученые, у этих рыбок возникновение разных вариантов гена привело к увеличению количества нейронов в головном мозге, однако число нервных рецепторов в кишечнике снизилось [6].
В своем исследовании зебровых амадин (птиц из семейства ткачиковых — прим. перев.) группа ученых из Техасского Юго-западного медицинского центра обнаружила, что при нокауте гена FOXP1 молодые птицы-самцы не могут разучивать песни взрослых [7]. FOXP1 участвует в патогенезе РАС, а нокаут этого гена связан с речевыми и когнитивными нарушениями. Эти исследования на животных моделях показывают, как провести параллель между генотипом и фенотипом. Однако Эйхлер предупреждает, что не все так однозначно. Эти связи выглядят проще, чем есть на самом деле. Он отмечает, что ген CHD8 — только один ген из 500 потенциальных. Хотя тесная связь с развитием РАС уже установлена у более чем десятка генов, в дальнейшем предстоит изучить еще десятки. Эти исследования также демонстрируют важность подхода к изучению РАС, который Эйхлер называет «приоритетом генотипа». Он пишет, что для излечения 15 пациентов с тяжелыми мутациями потребовалось точечное повторное секвенирование 6176 пациентов с аутизмом и задержкой развития. Поэтому повторный клинический контакт и подробная характеристика этой небольшой подгруппы оказалось критически важными [6].
Эти и другие исследования предоставляют ценную информацию об этиологии и механизмах РАС. Это потенциально важно для разработки терапевтических подходов. Однако есть и трудности. Это гетерогенность клинических симптомов, а также то, что только часть случаев среди всех РАС объясняются особенностями конкретного гена и, таким образом, потенциально поддаются молекулярной таргетной терапии. Сандерс надеется, что в будущем будут открыты общие закономерности развития аутизма (рис. 2). Он проводит параллель с ишемической болезнью сердца, при которой в качестве факторов риска выступают повышенный уровень холестерина, курение, артериальная гипертония, сахарный диабет. Однако все звенья патогенеза сходятся в процессе развития атеросклеротической бляшки в коронарной артерии. Ученого беспокоит то, что необходимо разработать сотни различных лекарственных средств для воздействия на каждый ген, и все равно эти средства окажутся эффективными только у 15 % людей с аутизмом, у которых присутствует уже выявленное генетическое заболевание.

Диагностика аутизма
По некоторым оценкам, только в США около миллиона пациентов с диагнозом РАС в возрасте от 5 до 17 лет. Даже сегментированный рынок диагностических тестов востребован, особенно тесты, выявляющие много подтипов РАС. Но постановка точного диагноза очень сложна, потому что симптомы многих нарушений развития нервной системы, таких как умственная отсталость, эпилепсия и расстройства речи, часто перекрываются или схожи между собой. Генетическое исследование — ключ к более точной диагностике даже до рассмотрения вопроса о характере лечения.
Но создание диагностической базы — сложная задача, поскольку за последние несколько лет наблюдается прорыв в открытии генов. Как заявляет Аманда Линди, руководитель отдела нейрогенетики в GeneDx — компании-разработчике диагностических средств (подразделение Национального Института Здравоохранения США до 2001 года), ученые ведут поиск новых вариантов генов по базам данных других исследований. GeneDx также проводит собственные исследования на пациентах, которые обращаются с целью диагностики.
Число пациентов с диагнозами РАС значительно увеличилось в последние годы, количество и виды доступных диагностических средств также возросло. Стивен Шерер, медицинский генетик из Педиатрической клиники в Торонто, провел систематический анализ средств диагностики РАС, присутствующих на рынке. Ученый обнаружил повсеместное распространение в отношении числа и уникальности вариантов. В обзоре коммерческих тестов РАС в 2018 году количество вариантов патологии, поддающихся идентификации, варьировалось от 26 до 2562. Согласно Шереру, в настоящее время генетическое тестирование обычно начинается с анализа с применением микрочипов, который позволяет выявить 7 % вариантов (в основном вариации числа копий генов).Если ничего не обнаружено, следует экзомное секвенирование. Оно выявляет еще 7 % неперекрывающихся вариантов — обычно более мелких генетических изменений, таких как точечные мутации или небольшие делеции. По его словам, в основном эти две методики дополняют друг друга. Хотя некоторые компании сообщают об экзомном секвенировании, на самом деле многие из них секвенируют весь геном, так как это более эффективно и способствует быстрому получению данных.
Ambry Genetics предлагает широкий спектр тестов для неврологических заболеваний, основанный на распознавании порядка 200 генов. По словам представителей компании, он охватывает более 60 % случаев обращений, при которых определяется генетическая причина таких нарушений психического развития, как задержка развития, умственная отсталость и/или РАС. Но они также предлагают более точную диагностику: при наличии РАС проводится проверка нескольких десятков генов. По словам Келли Радтке, управляющей отделом исследований редких заболеваний в Ambry, существует определенный интерес к созданию более специфичных панелей генов. Меньшие по размеру панели включают гены, которые считаются не просто связанными с РАС, но и служат их причиной. В базу по аутизму ученые включили гены, которые были выявлены только у пациентов с изолированными неврологическими нарушениями, такими как задержка развития или поведенческие расстройства. Однако гены, связанные с мультисистемными генетическими синдромами, у которых много дополнительных, часто очень характерных симптомов, не включались в данную панель.
Но существуют причины для охвата множества генов. Поскольку тесты недешевы, и не все пациенты и/или их родственники готовы платить за расширенный анализ, увеличение количества генетических вариантов, охватываемых одним тестом, повышает шансы что-либо выявить. По словам Шерер, компаниям невыгодно затрачивать производственные мощности на создание небольшой панели, в которой содержались бы гены, ответственные за развитие только 1–2 % случаев аутизма. Таким образом, в базу вносятся гены, характерные не только для аутизма, поэтому некоторые панели содержат тысячу или даже более генетических вариантов. Также в панель могут быть внесены генетические варианты, связанные с задержками развития нервной системы и ассоциированные с аутизмом гены, участвующие также в развитии некоторых менделевских заболеваний.
Например, панель GeneDx Autism/ID Xpanded включает более 2300 генов, около 400 из которых имеют известные патогенные варианты (в отличие от генов, просто связанных с развитием патологии). Линди сообщает об изучении более 10 000 пациентов, произведенном с помощью этой базы генетических последовательностей. Количество пациентов, которых исследователи тестируют с помощью этой панели, возрастает с каждым днем, равно как и число генов, патогенные варианты которых удалось выявить. Несмотря на то, что списки генов быстро устаревают (говорят, что в тот день, когда создается база секвенирования генов РАС, она уже устарела), базы, основанные на экзомных последовательностях и фенотипических особенностях обновлять быстрее и легче, что делает их наиболее актуальными на рынке.
И родители, и опекуны для получения ответов на вопросы, связанных с РАС, все чаще обращаются к генетическому тестированию. По словам Линди, число тестов для диагностики аутизма резко повысилось. Количество заказов на получение диагностических тестов Autism Xpanded выросло по сравнению с заказами на тесты для выявления других патологий не линейно, а экспоненциально.
В конечном счете, цель состоит в обнаружении генов, связанных с развитием характерных проявлений аутизма, таких как определенное поведение в обществе, тревожность или умственная отсталость. Шерер скептически относится к тому, что один единственный ген может определять такие признаки, как поведение в обществе. Тем не менее, эти предположения необходимо проверять. И есть повод для оптимизма. Линди заявляет, что для некоторых фенотипов, связанных с РАС, удалось найти гены, служащие причиной развития большинства случаев. Например, ученые обнаружили, что при эпилептическом фенотипе в 24,8 % положительных случаев (т. е. при обнаружении пробанда, несущего патогенный аллель гена) присутствовал патогенный вариант гена SCN1A, а в 13,2 % случаев — вариант гена KCNQ28.
Генная терапия моногенных заболеваний
Наиболее вероятно, что первыми мишенями для генной таргетной терапии служат высокопенетрантные моногенные состояния с признаками РАС. Синдром ломкой Х-хромосомы — частая форма умственной отсталости, связанная с аутизмом — можно выявить по частым повторам в промоторной области гена FMR1, кодирующего белок FMRP (с англ. дословно — «белок умственной отсталости, связанной с ломкой Х-хромосомой»). Это РНК-связывающий белок, играющий жизненно важную роль в синаптогенезе и синаптической пластичности, влияющий на различные аспекты метаболизма и биологии мРНК. Синдром Ретта, которым страдают 10 тыс. человек в Соединенных Штатах и характеризующийся многими общими с аутизмом признаками (включая потерю речи и компульсивное поведение), вызван мутациями в гене MECP2. Туберозный склероз, который приводит к аутизму почти у половины больных, вызван мутациями в двух генах, кодирующих белки туберозного склероза 1 и 2 (TSC1 и TSC2). Синдром Ангельмана, которым страдают около 15 000 человек в Соединенных Штатах, возникает в результате делеции материнской копии гена убиквитинлигазы E3A (UBE3A, дупликации которого также связаны с аутизмом).
Результаты доклинических исследований позволяют предположить несколько возможных путей воздействия на гены, связанные с развитием аутизма. Первый заключается в поддержании функции с помощью вирусной генной терапии. В качестве средства доставки генного материала в мозг широкое признание получили аденоассоциированные вирусы (ААВ), в частности, ААВ серотипа 9 (AAВ-9), обладающий уникальным свойством преодолевать гематоэнцефалический барьер (ГЭБ).
При применении генной терапии FMR1, опосредованной ААВ-9, на модели синдрома ломкой X хромосомы у мышей были получены обнадеживающие результаты, которые заключались в частичной или полной коррекции проявлений заболевания [9]. Однако наиболее выдающиеся успехи этой методики достигаются в случае синдрома Ретта.
После приобретения AveXis для лечения этого редкого нарушения психического развития, которое практически избирательно поражает девочек, компания Novartis ввела в клинику AVXS-201 — генный препарат на основе AAВ-9, направленный на ген MECP2. Доклинические исследования на мышах и приматах свидетельствуют, что данный метод генной терапии способен восстанавливать экспрессию белка MECP2 в головном мозге [10]. Компании пришлось перезапустить многие доклинические испытания после проведенного внутреннего расследования в 2019 году, согласно результатам которого исследователи AveXis совершали манипуляции с результатами при подготовке набора данных для другого генного препарата на основе AAV-9 — «Золгенсмы» (онасемноген абепарвовек; изначально — препарат для лечения спинальной мышечной атрофии. — Прим. перев.). Теперь Novartis планирует представить для клинических испытаний новое лекарственное средство (IND — от англ. investigational new drug) в конце 2021 года.
Sarepta Therapeutics — еще одна компания, в которой проводятся исследования генной терапии на основе AAВ при синдромах Ретта и Ангельмана. Sarepta сотрудничает со StrideBio по программам разработки ААВ-терапии для воздействия на гены UBE3A, SCN1A или MECP2. В компании StrideBio, основанной Мавис Агбандже-Маккенна из Университета Флориды и Аравиндом Асоканом из Университета Дьюка, стремятся использовать результаты криоэлектронной микроскопии высокого разрешения капсидов ААВ для создания векторов [11], способных уклоняться от нейтрализующих антител и обладающих повышенным тропизмом к тканям-мишеням, векторной эффективностью и технологичностью производства. Sarepta объединилась с учеными из Массачусетского университета (Гуанпин Гао, Мигелем Сена Эстевесом и Майклом Грином) для исследования синдрома Ретта (за развитие которого отвечает ген MECP2), а также для создания библиотеки новых капсидов ААВ (полученных из клеток человека).
Наконец, PTC Therapeutics разрабатывает AGILAS — генный препарат, доставляемый в гиппокамп, на основе AAВ-9 — для воздействия на UBE3A при синдроме Ангельмана, которому Управление по контролю качества продуктов питания и лекарственных средств США (FDA) присвоило статус орфанного заболевания в 2015 году. Компания исследует несколько других серотипов AAВ; в настоящее время подача заявки на статус IND в GT-AS отложена вследствие пандемии COVID-19, однако процесс готовы возобновить в конце этого года. Также Amicus Therapeutics предоставила лаборатории Джеймса М. Уилсона (директор Программы генной терапии и Центра лечения орфанных заболеваний Университета Пенсильвании) 50 млн долларов для доработки и улучшения вектора из группы AAВ-9 с целью обеспечения его доставки в мозг [12]. Цель программы — поиск человеческих аналогов гликозилфосфатидилинозитол-заякоренного белка локуса А лимфоцитарного комплекса 6 — белка, обнаруженного в ГЭБ мышей, — для облегчения проникновения [вектора] в ткань мозга.
Помимо традиционной генной терапии AAV несколько компаний используют другие таргетные молекулярные подходы к лечению РАС, такие как применение препаратов из группы АСО (антисмысловых олигонуклеотидов) малых молекул, которые обеспечивают считывание связанных с заболеванием нонсенс-мутаций, а также метод редактирования генов на основе CRISPR.
Воздействие на РНК
Используя технологию, разработанную в лаборатории Linyan Meng из Бэйлорского университета в сотрудничестве с компанией Ionis Pharmaceuticals (впоследствии переименована в Isis Pharmaceuticals) [13], Фонд по лечению синдрома Ангельмана (FAST) создал дочернюю компанию GeneTx Biotherapeutics, которая разрабатывает GTX-102 — 2'- метоксиэтил(MOE)-гэпмер АСО. Гэпмеры — короткие антисмысловые олигонуклеотидные последовательности ДНК, по краям которых находятся РНК-подобные фрагменты. — Прим. перев. Гэпмер GTX-102 реактивирует отцовский аллель гена UBE3A. Его экспрессия подавляется после связывания с естественной антисмысловой последовательностью — транскриптом UBE3A-AS. Таким образом, транскрипция останавливается. Данный метод лечения восстанавливает считывание нижележащего гена UBE3A с противоположной стороны.
В декабре 2020 года на ежегодном международном саммите FASТ были представлены промежуточные результаты фазы I/II клинического испытания (NCT04259281), в котором участвовали пять пациентов. Согласно этим результатам, концентрация GTX-102 в плазме оказалась пропорциональна дозе, при которой наблюдалось среднее изменение +2,4 (т. е. улучшение) по общей шкале оценки клинического состояния при синдроме Ангельмана (CGI-I-AS). У всех пациентов было обнаружено улучшение состояния и стабильный уровень контроля приступов. Для четырех из пяти пациентов, получавших лечение, данные электроэнцефалограммы, которые были получены слепым методом в начале эксперимента и на 128-й день, свидетельствовали о снижении частоты встречаемости аномалий (эпилептиформной активности), характерных для пациентов с синдромом Ангельмана. Ожидается, что исследование возобновится в ближайшие месяцы. В то же время компания Roche Pharmaceuticals (расположенная в Базеле, Швейцария) ведет разработку RO-7248824 — препарата АСО на основе заблокированной нуклеиновой кислоты (ЗНК), направленной на родительскую антисмысловую РНК гена UBE3A-AS у пациентов с синдромом Ангельмана. В июне 2020 года в Европе и США стартовала I фаза открытого многоцентрового исследования с повышением дозы для изучения безопасности, переносимости, фармакокинетики и фармакодинамики препарата RO-7248824 у пациентов в возрасте до 18 лет.
В 2018 году компания Biogen также подписала соглашение о сотрудничестве с Ionis для разработки проекта 2'-MOE-гэпмеров АСО с целью лечения различных неврологических расстройств. При этом компании Ionis было выплачено 375 млн долларов авансовых платежей и 500 миллионов долларов капитала плюс 125 млн долларов денежной премии. В число болезней, на которых хотят сосредоточиться в Biogen, — синдром ломкой X-хромосомы, синдромы Ангельмана и Ретта.
Наконец, PTC Therapeutics приступила к фазе II перекрестного исследования своего препарата на основе малых молекул — «Трансларны» (аталурен) — направленного на РНК. Этот препарат в 2016 году был одобрен в Европе для терапии мышечной дистрофии Дюшенна, опосредованной нонсенс-мутациями, а также для лечения лекарственно-устойчивой эпилепсии у пациентов с нонсенс-мутациями при состоянии, близком к аутизму (т. н. расстройстве с дефицитом белка CDKL5). Существуют разногласия по поводу способности «Трансларны» проникать через ГЭБ. Однако согласно результатам исследования, доказывающим принцип действия препарата, при более высоких дозах это возможно [14].
Редактирование и связывание генов
Большинство компаний, связанных с редактированием генов, занимаются лечением заболеваний, в патогенезе которых задействованы ткани и клетки, легко поддающиеся внесению генов извне. В число таких тканей входят глаза, клетки крови или печень. Поскольку при РАС необходимо введение генетического материала в ткань мозга, выбор падает на прямую инъекцию вектора AAВ-9 в большую цистерну мозга. При этом экспрессия необходимых генов наблюдается в лучшем случае в ~ 10 % целевых нейронов. Исследователи Центра медицинских наук Техасского университета и Калифорнийского университета в Беркли провели совместное изучение стартапа GenEdit в области генной терапии. Ученые исследовали введение однократной дозы компонентов CRISPR, связанных с наночастицами золота (конъюгированные с олигонуклеотидами), которые затем покрывались полимерной оболочкой, разрушающейся в эндосомах [15]. Введение компонентов в полосатое тело мышей с синдромом ломкой Х-хромосомы, у которых наблюдалась низкая экспрессия гена Fmr1, в значительной степени спасала животных от формирования поведенческих расстройств и проявлений повторяющегося поведения.
Исследовательская группа, возглавляемая Ниссимом Бенвенисти из Еврейского университета в Иерусалиме и Донг-Вуком Кимом из Медицинского колледжа Университета Йонсей в Сеуле, обнаружила, что редактирование генома работает в человеческих клетках — потомках клеток лиц с синдромом ломкой Х-хромосомы. Используя эмбриональные стволовые клетки и индуцированные плюрипотентные стволовые клетки, полученные от больных, ученым удалось показать, что коррекция повторов CGG (цитозин-гуанин-гуанин) приводит к деметилированию вышележащего CpG-островка промотора гена FMR1. Это способствовало формированию открытого состояния хроматина и инициации транскрипции [16].
Сотрудничество между Novartis и Sangamo в июле прошлого года открыло возможность для развития еще одного инструмента редактирования генов — ZFP-факторов транскрипции (от англ. Zinc finger protein transcription factors — транскрипционные факторы, содержащие цинковые пальцы; ZFP-TF). На данный момент препараты на их основе еще не проходят клинические испытания (см. вставку 1). По словам Гопи Шанкера, руководителя отдела психиатрии и временного руководителя отдела нейробиологии Института биомедицинских исследований Novartis, ZFP-TF служат дополнением к другим разработкам компании в области генной терапии на основе AAВ-9. В настоящее время Novartis занята продажей патента на препарат «Золгенсма» на основе AAВ-9, одобренного для лечения спинальной мышечной атрофии в 2019 году. Именно данное обстоятельство стало основанием для приобретения AveXis за 8,7 млрд долларов в 2018 году.
Вставка 1 | От редактирования генов к регуляции их работы
По словам Гопи Шанкера, директора отдела психиатрии и временного руководителя отдела нейробиологии Института биомедицинских исследований Novartis, применение компанией технологии Sangamo предлагает широкий спектр заболеваний ЦНС в качестве потенциальных мишеней. Некоторые методы терапевтического воздействия включают не только простое редактирование генов.
В последнем рецензируемом исследовании компании сочетается ДНК-связывающая специфичность ZFP с репрессивной активностью TF для создания селективного в отношении аллелей регулятора транскрипции [22]. Авторы изучали активность набора факторов подавления транскрипции ZFP-KRAB, разработанных против CAG-повторов, содержащихся в смысловой и антисмысловой последовательностях в фибробластах и нейронах пациентов с болезнью Хантингтона (БХ). Наиболее эффективные домены ZFP-KRAB селективно подавляли более 99 % аллелей гена хантингтина (HTT), который вызывает БХ. При этом сохранялась экспрессия более 86 % нормальных аллелей HTT. По словам авторов, ZFP-TF, доставляемые с помощью лентивирусного вектора или AAВ-6, оставались активными в нейронах более 100 дней в культуре клеток и в течение девяти месяцев — в мозге мышей. На трех мышиных моделях БХ было показано улучшение молекулярных, гистопатологических, электрофизиологических и функциональных конечных точек.
Еще одним ключом к этому сотрудничеству служит утверждение представителей компании Novartis о том, что они нашли новые интересные мишени воздействия, но какие — пока не разглашают. Рикардо Долметч, руководитель отдела нейробиологических исследований в Novartis, сообщает, что методы воздействия на наиболее распространенные мутации либо уже находятся в стадии разработки, либо очень конкурентоспособны. Компания Novartis и другие заняты поиском вариантов генов, ответственных не просто за развитие единичных симптомов — например, таких как эпилепсия или расстройства ЖКТ, а за целый комплекс состояний — как это происходит в случае аутизма.
Как поясняет Шанкер, у этой технологической платформы есть преимущество: существует ограничение на размер гена, который возможно доставить в составе вектора ААВ. С помощью ZFP можно использовать генную терапию для регуляции экспрессии генов, усиливая или ослабляя ее вместо того, чтобы заменять или редактировать их структуру. ZFP позволяет достичь этого независимо от размера гена-мишени. ZFP-TF встраиваются в вектор AAВ и доставляются в ЦНС, а затем специфически связываются с геном-мишенью или его аллелем без интеграции в геном.
Возникающие трудности
Несмотря на то, что работа с моногенными патологиями повышает уверенность исследователей в том, что воздействие на мишень приведет к смягчению протекания заболевания, неудачи в разработке лекарств неизбежны. В 2014 году компания Novartis прекратила производство мавоглуранта (AFQ056) — низкомолекулярного селективного антагониста метаботропного рецептора глутамата 5 (mGluR5) — после того, как препарат потерпел неудачу в двух 12-недельных плацебо-контролируемых испытаниях фазы IIB у подростков и взрослых пациентов. В отличие от большинства других прошедших испытания симптоматических методов лечения при синдроме хрупкой Х-хромосомы, мавоглюрант ингибирует активацию рецептора mGluR5, тем самым снижая выраженность дефектов синапсов. Это прямое следствие отсутствия белка FMRP. В конце прошлого года произошел аналогичный случай, в ходе которого компания Ovid Therapeutics потерпела сокрушительный провал фазы III испытания своего таргетного низкомолекулярного препарата габоксадола для лечения синдрома Ангельмана. Габоксадол — агонист δ-селективного рецептора γ-аминомасляной кислоты A (GABAA), ген которого подвергается делеции вместе с геном UBA3.
Конечно, препараты на основе малых молекул — это испытанный метод лечения. Генная терапия, напротив, сопряжена с определенными рисками, в том числе ожидаемым истощением новых генетических мишеней со временем. Хотя в целом векторы AAВ считаются безопасными — отчасти потому, что они являются эписомальными структурами (т. е. не интегрируются в геном), — результаты длительного исследования собак, которых лечили от гемофилии А, позволяют предположить, что это не всегда так. В ходе изучения собак, которым был введен ген собачьего VIII фактора свертывания крови в составе векторов AAВ, проводимого в течение десяти лет под руководством Джанга Нгуена из Пенсильванского университета, были обнаружены доказательства того, что встраивание векторов происходит преимущественно в областях генома, связанных с канцерогенезом, что увеличивает риск развития долгосрочных неблагоприятных последствий.
Еще одной проблемой безопасности генной терапии, ожидаемой в доклинических исследованиях, служит токсичность в отношении спинальных ганглиев (СГ). СГ — это скопление нервных клеток за пределами спинного мозга, отвечающее за передачу сенсорных сигналов. Токсичность в отношении СГ была впервые обнаружена в исследованиях на приматах с использованием векторов AAВ для введения генов по спинномозговой жидкости и внутривенно. Также она наблюдалась у свиней [12, 17]. В этих исследованиях сообщалось о развитии дегенерации аксонов в некоторых проводящих путях спинного мозга и периферических нервах, что, как предполагается, связано со гиперэкспрессией трансгена.
Возможно, Novartis уже столкнулась с данной проблемой. FDA настаивают на проведении нового клинического испытания препарата Zolgensma, так как в испытаниях на животных, выполненных первоначальным разработчиком лекарственного средства — компанией AveXis — обнаружилось воспаление в СГ с участием мононуклеаров. Novartis сможет получить одобрение на данный препарат в США только после повторных испытаний. Агентство занято поиском данных, подтверждающих результаты исследования STRONG компании Novartis, в котором изучался вариант «Золгенсмы» для спинномозгового введения у детей старшего возраста, страдающих спинальной мышечной атрофией. В настоящее время препарат «Золгенсма» одобрен для внутривенного введения детям в возрасте до двух лет с более серьезными заболеваниями. Однако Novartis не сможет начать новое испытание в Соединенных Штатах до тех пор, пока FDA не снимет запрет на клинические исследования, который был наложен в прошлом году из-за зафиксированных признаков воспаления, наблюдаемых на животных моделях.
На основе результатов метаанализа, опубликованного летом 2020 года, можно предположить, что токсичность в отношении СГ может быть обычным побочным явлением у приматов и зависит от пути введения (данное явление наблюдается чаще при введении лекарственных средств интратекально в спинномозговую жидкость, чем при внутривенной инъекции), возраста и дозы, однако может и не проявляться клинически [12]. Авторы собрали данные 33 доклинических испытаний на более чем 250 приматах и провели сравнение нескольких факторов, включая пути введения, векторный капсид и трансген. Согласно заявлению Уилсона, который был соавтором исследования, признаки патологических изменений в СГ становятся почти повсеместными после введения векторов ААВ в спинномозговую жидкость приматов. Однако ни у одного из животных, которые получали векторы, экспрессирующие терапевтический трансген, не было выявлено каких-либо клинических проявлений.
Нецелевое воздействие также следует учитывать при использовании терапии на основе АСО и редактирования генов, несмотря на их специфичность к определенным последовательностям. С развитием технологий обнаружения нецелевых эффектов редактирования генов станет понятнее, насколько серьезна данная проблема и какие могут быть варианты ее решений. В нескольких недавних исследованиях путем изучения полного транскриптома культуры клеток человека после воздействия терапии с применением АСО различной длины были отмечены нецелевые эффекты [18, 19]. Также после первоначального ажиотажа научного сообщества относительно возможности действия на мишень с помощью белков с цинковыми пальцами появились сообщения о нецелевой инцизии [вырезании] генов [20], хотя исследователи из Sangamo недавно сообщили, что в скором времени решение может быть найдено [21].
Возможный повод для оптимизма
Остается много вопросов о наилучших способах поиска и изучения мишеней для лечения заболеваний, связанных с нарушениями развития нервной системы, таких как РАС. Тем не менее, похоже, что генная терапия на основе векторов только набирает обороты, что может помочь решить проблему нецелевого редактирования. В настоящее время проводятся десятки клинических испытаний генной терапии. Так, в 2020 году было объявлено о нескольких соглашениях между крупными фармацевтическими компаниями и компаниями, занимающимися генной терапией: Bayer, Eli Lilly и UCB присоединились к Novartis с крупными инвестициями в эту область. В этом году начнется несколько клинических испытаний таргетных препаратов для лечения РАС.
Учитывая тысячи генетических нарушений, лежащих в основе развития РАС, которые возникают в результате разнообразных мутаций — от простых точечных до делеций хромосомных областей, охватывающих многие гены, необходимы различные методы молекулярной таргетной терапии. На данный момент та область, где разработка лекарств для массовой продажи возможна и целесообразна — это моногенные расстройства. Сандерс выражает свое беспокойство по поводу того, что необходимо разработать сотни различных лекарственных средств, каждое из которых должно воздействовать на определенный ген. При этом эффект может быть достигнут только у 15 % страдающих аутизмом людей с выявленным генетическим нарушением.
Кроме того, необходимы дальнейшие исследования в области диагностики различных вариантов РАС, прежде чем серьезно рассматривать возможности массового применения генной терапии. По словам Шерера, так как аутизм не является смертельным заболеванием, принятие радикального решения и испытание его на любых стадиях является очень сложным процессом. Также он считает, что обсуждение возможностей применения генной терапии в данный момент реально лишь в области исследований и может использоваться в качестве метода для создания моделей, в которых изучались бы способы коррекции мутаций.
