
Мозг и кислород: в бесконечной пляске смерти

Человеческий мозг весит в среднем 1400 г, однако расходует около 20 % от общего потребляемого организмом кислорода: это уходит на поддержку ~ 86 млрд нейронов и их невообразимо сложного коннектома, включающего в себя триллионы синапсов [1–3], что в свою очередь обслуживается обслуживается 250–300 млрд глиальных клеток [4,5].
Такое интенсивное «дыхание» необходимо мозгу для процессов мышления, и даже транзиторная ишемия приводит к массивной нейродегенерации [6]. Без достаточного количества кислорода митохондрии будут не в состоянии использовать кислород в дыхательной цепи, обеспечивая синтез АТФ, поэтому даже 30-минутная гипоксия мозга при ишемическом инсульте приводит к катастрофическим последствиям — гибели около 1,9 млн нейронов и 14 млн синапсов ежеминутно [6,7]. Тем ни менее, как это ни парадоксально на первый взгляд, мозг так тщательно регулирует потребление кислорода по той простой причине, что молекулярный кислород в основном состоянии является бирадикалом и, следовательно, потенциально токсичным, мутагенным газом.
К счастью, окислительный потенциал кислорода сдерживается одной квантово-химической тонкостью — его два электрона имеют параллельные спины, и одновременно кислород может принять лишь один электрон [8,9]. Если это свойство ограничивает реактивность кислорода, тогда почему он считается токсичным? Ответ заключается в способности кислорода образовывать свободные радикалы и другие активные формы кислорода (АФК), а именно супероксидный анион-радикал (O2•–), перекись водорода (H2O2) и гидроксил-радикал (OH•) (их биохимия рассмотрена в работах [8,10,11]). Такие соединения обычно считаются «темной стороной» биохимии кислорода — неизбежной платой за использование кислорода для дыхания [12].
Долгое время предполагалось, что их неконтролируемый и нежелательный синтез повышает чувствительность мозга к окислительному стрессу. Действительно, окислительный стресс тесно связан с нейродегенерацией [13,14]. Однако простое разделение кислорода на «хороший» — молекулярный и его «плохие» реактивные формы (к примеру, O2•–) не может объяснить причину и механизм высокой чувствительности мозга к окислительному стрессу, потому как такой подход неверен.
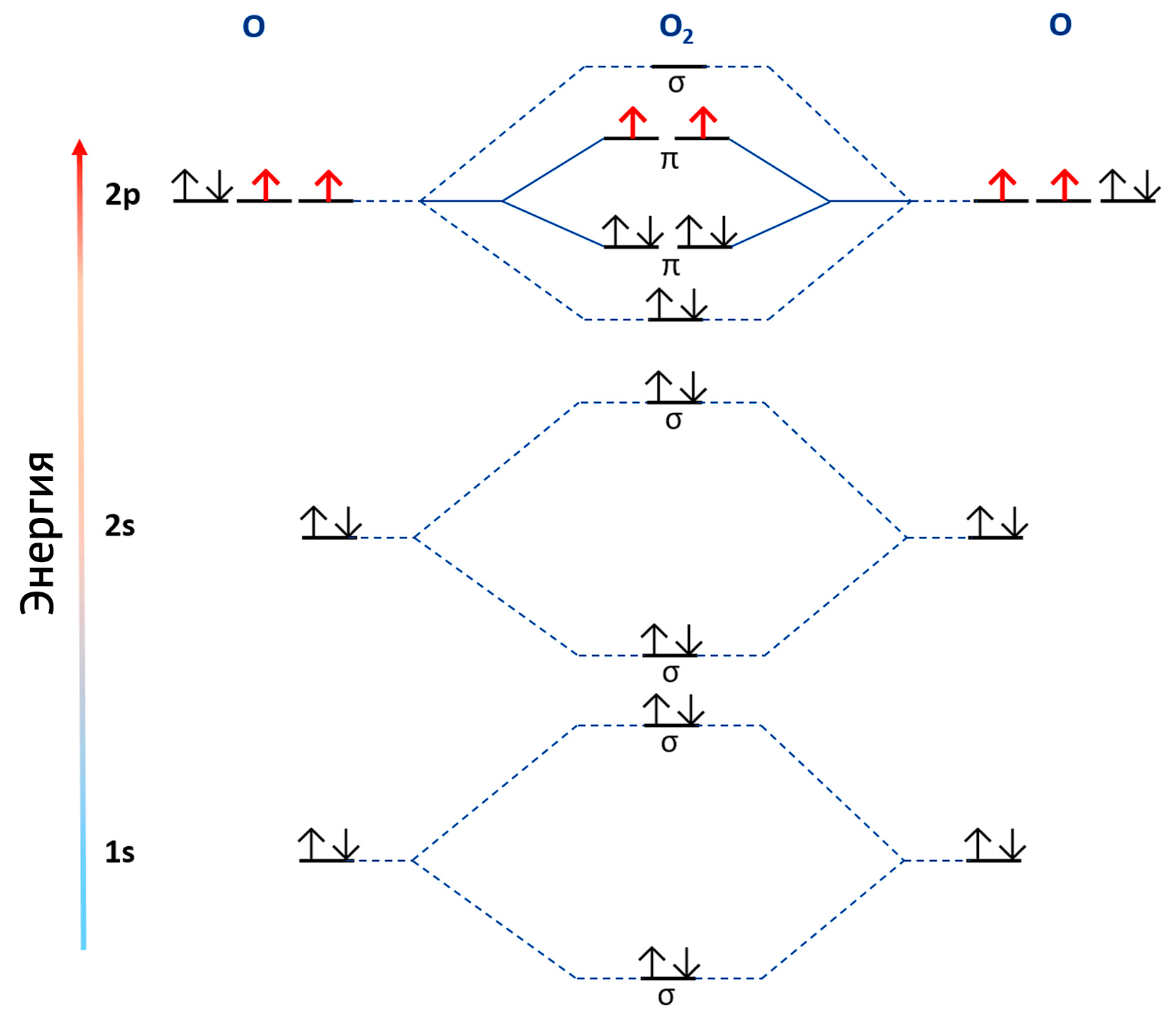
3∑g–O2 является двухвалентным радикалом, так как его одиночные электроны располагаются на двух вырожденных антисвязывающих π*2p орбиталях (показаны красным) и имеют параллельные спины, что определяет спин-поляризованное состояние 3∑g–O2. Спиновое ограничение в данном случае возникает весьма удачно, так как оно сдерживает реактивность 3∑g–O2.
Чтобы понять, почему и как мозг восприимчив к окислительному стрессу, надо отказаться от догмы о том, что АФК априори являются лишь токсичными метаболическими продуктами, и учесть некоторые их особенности. Например, важно помнить о том, что именно высокая чувствительность мозга к гипоксии является причиной возникновения адаптивных митохондриальных сигналов в ответ на воздействие O2•– [7]. Такой «редокс-сигналинг» вовсе не является исключением, а, напротив, широко распространен [15,16]. Окислительный стресс может возникать, когда редокс-сигналинг нарушается («двуликость» редокс-сигналинга).
Чувствительность мозга к окислительному стрессу редко подвергалась глубокому анализу, поэтому попытки устранить сложную взаимосвязь между окислительным стрессом и нейродегенерацией до сих пор не увенчались успехом. Для того чтобы лучше понять механизмы этих процессов, авторы выделили 13 биохимических причин уязвимости мозга к окислительному стрессу. Для этого они опирались на плодотворную работу Барри Холливелла и Джона Гуттериджа [17–19].
1. Редокс-сигналинг: активные формы кислорода выполняют важные биологические функции
Единственная и часто недооцененная причина, по которой мозг восприимчив к окислительному стрессу, заключается в том, что активные формы кислорода играют важную роль в функционировании организма [19,20]. Это иллюстрируют 2 примера.
Во-первых, группой ученых во главе с Чанг [21] было доказано, что O2• – и H2O2, синтезируемые НАДФH-оксидазой 2 (NOX2), регулируют рост клеток-предшественников гиппокампа через сигнальный путь PI3K/Akt. Их исследования выявили благоприятное влияние O2• –/H2O2 на поддержание гомеостаза жизненно важных нейрональных предшественников [21]. Экспрессия NOX2 — важного фермента, необходимого для синтеза O2• – , сама по себе указывает на существенную роль редокс-сигналинга [22,23]. Соответственно, можно сделать вывод, что изоформы NOX регулируют долговременную потенциацию в гиппокампе — важный процесс, необходимый для памяти и обучения [24]. Делеция по гену NOX2 приводит к когнитивным нарушениям у мышей [25].
Во-вторых, группа ученых под руководством Vriz установила положительное влияние генерируемой NOX H2O2 на аксональное наведение и регенерацию [26,27]. Аксональное наведение прокладывает нервные пути в развивающемся мозге частично с помощью секреции хемоаттрактантов и хемореппелентов, обеспечивающих правильную иннервацию [28]. Фармакологическое подавление активности NOX2, а значит и синтеза этим ферментом O2• –/H2O2 замедляло рост аксонов ганглионарных клеток сетчатки у зародышей рыбок данио-рерио, что свидетельствует о выполняемой H2O2 функции эндогенного хемоаттрактанта [26].
2. Кальций
Потенциал действия вызывает увеличение количества поступающих ионов Ca2+ в пресинаптических терминалях, на 4 порядка увеличивая их концентрацию (с 0,01 до ~ 100 мкмоль на литр [29]). Вход ионов Ca2+ вызывает экзоцитоз гранул нейромедиаторов [29]. Следовательно, потенциал-зависимые токи кальция контролируют синаптическую пластичность в обоих направлениях [30]. Такая двухсторонняя синаптическая пластичность является фундаментальным свойством, необходимым для функционирования мозга. В качестве хорошего примера таких функций можно привести в пример процессы обучения и памяти [31–33]. Зависимость работы мозга от сигналинга Cа2+ [34] может вызывать окислительный стресс, природа которого, в свою очередь, зависит от условий среды ввиду сложных взаимоотношений ионов кальция с окислительно-восстановительными процессами [19]. Заинтересованному читателю рекомендуется ознакомиться с исчерпывающим обзором взаимоотношений Са2+/редокс [35], а авторы ограничатся тремя моментами.
Во-первых, входящий ток кальция активирует нейрональную NO-синтазу (nNOS), что в условиях достаточного количества O2 и НАДФH [37] повышает интенсивность синтеза оксида азота I (NО). Поэтому остающийся повышенным внутриклеточный уровень Са2+ может увеличивать концентрацию NO, который имеет свойство угнетать работу митохондрий, связываясь с цитохром с-оксидазой (COX) (комплекс IV — прим. перев.) [38]. По мере диффузии NO реагирует с O2• –, в результате чего образуется пероксинитрит (ONOO–) [39]. Пероксинитрит может реагировать с CO2, образуя нитрозопероксикарбонат (ONOOCO2–), который распадается до радикалов карбоната (CO3• –) и диоксида азота (NO2• –) [40]. CO3• – и NO2• – могут способствовать нейродегенерации, например, путем нитрования белка теплового шока 90, и таким образом вызывать апоптоз нейронов, как это происходит при развитии бокового амиотрофического склероза (БАС) [41]. Сходный результат дает увеличение активности фосфолипазы А2 ионами Са2+ [34]. Изоформы фосфолипазы А2 расщепляют эфирные связи мембранных фосфолипидов, что может вызывать ферментативное (т. е. через ЛО [42]) и неферментативное перекисное окисление бисаллильных ненасыщенных липидов [43].
Во-вторых, рост внутриклеточной концентрации Са2+, обеспечивающий синаптическую пластичность [44], является также редокс-регулируемым [45,46]. Например, группа под руководством Хажноцки показала [47], что митохондриальные нанодомены H2O2 регулируют кальциевые токи. Приток кальция способствует формированию контактов между митохондриями и ЭПР, что получило название митохондрия-ассоциированных мембран ЭПР (mitochondria-associated ER membranes (MAM)) [48,49], что в итоге приводит к поглощению ионов кальция митохондриями. Рост концентрации ионов кальция в митохондриях, в свою очередь, стимулирует их высвобождение из ЭПР через усиление захвата ионов калия, в конечном итоге увеличивая объем матрикса и сжимая межмембранное пространство, в результате чего повышается концентрация H2O2 в MAM [47].
Авторы предполагают, что H2O2 вызывает высвобождение Ca2+ через рецепторы ИФ3 в соответствии с их редокс-регуляцией путем окисления цистеина [50]. Так как МАМ регулируют множество функций митохондрий (например, транспорт и биогенез [48]), можно легко представить, как нарушение регуляции внутри органелл будет вызывать аномальный синалинг Ca2+/H2O2, связанный с окислительным стрессом [45]. Для уверенности стоит добавить, что нарушение регуляции сигналинга МАМ связано с нейродегенерацией при болезни Альцгеймера и боковом амиотрофическом склерозе (БАС) [51]. Например, Стойка с соавт. [52] показали, что мутантный TD43 — патологический «триггер» при БАС и лобно-височной деменции [53] — уменьшает число контактов в МАМ и таким образом нарушает кальциевый гомеостаз (рис. 1–6).
Третья точка взаимодействия: избыток Са2+ в митохондриях приводит к открытию митохондриальных пор переходной проницаемости (mPTP) [54]. Открытие mPTP вызывает выход из митохондрий O2• –/H2O2 и нарушает синтез АТФ [55–57]. Временное открытие mPTP заставляет митохондрии восстанавливать уровень кальция в матриксе [54,58] и, скорее всего, запускает редокс-сигналинг посредством выхода O2• –/H2O2 из метаболизма матрикса митохондрий [59] (явление, которое может быть связано со сжатием митохондрий [60,61]). Длительное открытие mPTP является предвестником некроптоза [62]. Кроме того, избыток Са2+ может запускать апоптоз по внутреннему пути. Важно то, что и апоптоз, и некроптоз связаны с нейродегенерацией [63,64]. Так как поглощение Са2+ митохондриями является необходимым для поддержания синтеза АТФ [65–67], снижение концентрации кальция в митохондриях может запускать окислительный стресс, повышая концентрацию NADH и сопутствующий синтез O2• – FMN-сайтом комплекса I [68,69]. Кроме того, цитохром c может также запускать апоптоз, используя H2O2 для окисления кардиолипина, очень важного фосфолипида внутренней мембраны [70,71]. Неудивительно, что (1) мозг расходует так много энергии АТФ для поддержания кальциевого гомеостаза, и (2) нейродегенеративные заболевания обычно связаны с нарушением гомеостаза Са2+.
{kind=link}
3. Глутамат
Чрезмерное действие глутамата, например, через NMDA-рецепторы (NMDAR) вызывает эксайтотоксичность [72,73], которая следует за нарушением сигналинга Ca2+ (например, вызывая устойчивое действие кальпаина) [74]. Глутаматная эксайтотоксичность приводит к перегрузке кальцием митохондрий, высвобождению ими O2• –/H2O2, и в итоге клеточной смерти, обычно через апоптоз или некроз [17,75,76]. Приток Ca2+ может активировать nNOS, что позволяет NO• ингибировать ЦОГ, повышая тем самым концентрацию O2• –/H2O2/ONOO– в митохондриях. Согласно этому, фармакологическое ингибирование nNOS будет защищать от эксайтотоксичности [77]. А гибель нейронов по некротическому пути будет усиливать эксайтотоксичность, так как будет повышаться внеклеточная концентрация глутамата [78].
Интересно то, что действие глутамата на NMDAR может зависеть от пространственного расположения рецепторов: внесинаптическое действие глутамата будет вызывать эксайтотоксичность, в то время как активация синаптических NMDAR будет запускать адаптивный ответ [79–82]. Группа под руководством Hardingham показала [79], что активация синаптических NMDAR вызывает повышение экспрессии системы ферментов пероксиредоксин-тиоредоксин (PRDX-TRDX) и даунрегуляцию сигналов апоптоза. Возможно, в основе пространственной специфичности лежит избирательная работа ферментов: экстрасинаптические NMDAR, связанные с митохондриальным синтезом O2• –, вызывают нейродегенерацию, в то время как синаптические NMDAR, связанные с NOX2, способствуют протективным эффектам O2• – [83].
Кроме действия на рецепторы, глутамат может также вызывать эксайтотоксичноть, ингибируя систему транспортеров xС– [84], обменивающих внутриклеточный глутамат на внеклеточный цистин [85]. В клетке цистин восстанавливается до цистеина, который может быть использован глутамат-цистеин лигазой для синтеза глутатиона de novo [86]. Ингибирование транспорта цистина внутрь клетки вызывает истощение ресурсов глутатиона, что приводит к развитию окислительного стресса [84,87]. Снижение концентрации глутатиона также необходимо для запуска ферроптоза — варианта клеточной гибели, происходящего из-за окисления железа и перекисного окисления липидов [88], что подтверждает роль внеклеточного глутамата как сигнала запуска ферроптоза [89,90]. Однако как предостерегают Цао и Диксон [90], несмотря на общие моменты в окислительно-восстановительных процессах, глутамат-ассоциированная эксайтотоксичность, связанная с клеточной гибелью и ферроптозом, имеет различия, такие как вовлечение апоптотических сигналов в первом случае.
4. Глюкоза
Для поддержания работы нейронов человеческий мозг потребляет ~ 25 % глюкозы крови, что соответствует ~ 5,6 мг глюкозы на 100 г ткани мозга в минуту. Судьба глюкозы в головном мозге сложна и включает так называемое метаболическое сопряжение между нейронами и глией (рассмотрено в [3,92]). Суть его заключается в том, что сначала глия превращает глюкозу в лактат, после чего тот захватывается нейронами, превращается в пируват и окисляется в митохондриях чтобы обеспечивать клетки АТФ [3].
Это согласуется с данными, подтверждающими способность нейронов эффективно метаболизировать лактат [93,94]. Следствием этого является поддержание нейронами низкой активности фосфофруктокиназы (ФФК) — скорость-лимитирующего фермента гликолиза, чтобы использовать глюкозу преимущественно в пентозофосфатном пути (ПФП) [95]. С точки зрения окислительно-восстановительных процессов кажется, что межклеточное метаболическое сопряжение призвано компенсировать ограниченные способности нейронов метаболизировать карбонилы из-за низкой экспрессии глиоксилазы 1 (GLO1) и глиоксилазы 2 (GLO2) [96]. Обе изоформы глиоксилазы метаболизируют метилглиоксаль (МГ) [97] — потенциально токсичный дикарбонил, продукт триозофосфатизомеразы [98,99]. Скорость этого процесса регулируется количеством имеющегося глутатиона. GLO1 превращает связанный с глутатионом МГ (т. е. полутиоацеталь) в S-D-лактоилглутатион, после чего GLO2 превращает S-D-лактоилглутатион в D-лактат и глутатион. Низкое содержание изоформ GLO вкупе с относительно низкой концентрацией глутатитона делает нейроны уязвимыми к токсическим эффектам МГ: концентрации МГ в 250 µM достаточно для насыщения метаболических систем нейрона, в то время как метаболизму астроцитов для насыщения требуется 2 мM МГ [96], хотя обычно концентрация «свободного» МГ составляет 2–4 мкм [100]. Тем не менее, МГ в 50000 раз более реактивен, чем глюкоза, и может быстро приводить к образованию оснований Шиффа, способных гликировать (неферментативно гликозилировать) белки, РНК и ДНК [100].
В частности, гликирование белков приводит к формированию конечных продуктов гликирования (КПГ), которые могут вызывать окислительный стресс, активируя соответствующие рецепторы и запуская воспаление, что в итоге нарушает функции митохондрий и других белков [100–102]. КПГ могут синтезироваться и в отсутствие высокой интенсивности гликолиза, так как перекисное окисление липидов тоже может приводит к формированию МГ [101]. В итоге мозг оказывается уязвимым к окислительному стрессу, вызываемому глюкозой [97].
5. Митохондрии
Чтобы справиться с экстраординарными потребностями в АТФ, мозг потребляет несоразмерные количества O2 для поддержания окислительного фосфорилирования [3]. Нейроны тратят энергию АТФ на поддержание высоких биохимических градиентов и синаптической активности [103,104]. Чистые энергетические затраты на работу синапсов можно представить в виде затрат на высвобождение везикул с медиаторами, что составляет 1,64 × 105 AТФ в секунду на 1 везикулу [104,105]. Для удовлетворения таких энергетических потребностей нейронов и необходимы митохондрии, а ввиду ограниченной скорости диффузии АТФ особую важность представляют синаптические митохондрии [106]. Так как нейроны постоянно ограничивают активность ФФК, то есть скорость гликолиза [95], они очень сильно зависят от работы митохондрий, хотя синтез АТФ в синапсах могут некоторое время поддерживать гликосомы [107].
Кроме окислительного фосфорилирования, митохондрии являются важными узлами сигнальных путей, регулирующими множество жизненно важных процессов, начиная от гомеостаза Ca2+ и синтеза кластера Fe-S и заканчивая определением судьбы клетки [55,108,109]. Митохондрии нейронов это своего рода обоюдоострый меч: снабжая нейроны АТФ и сигналосомами, они при нарушении своей функции повышают уязвимость к нейродегенерации [110].
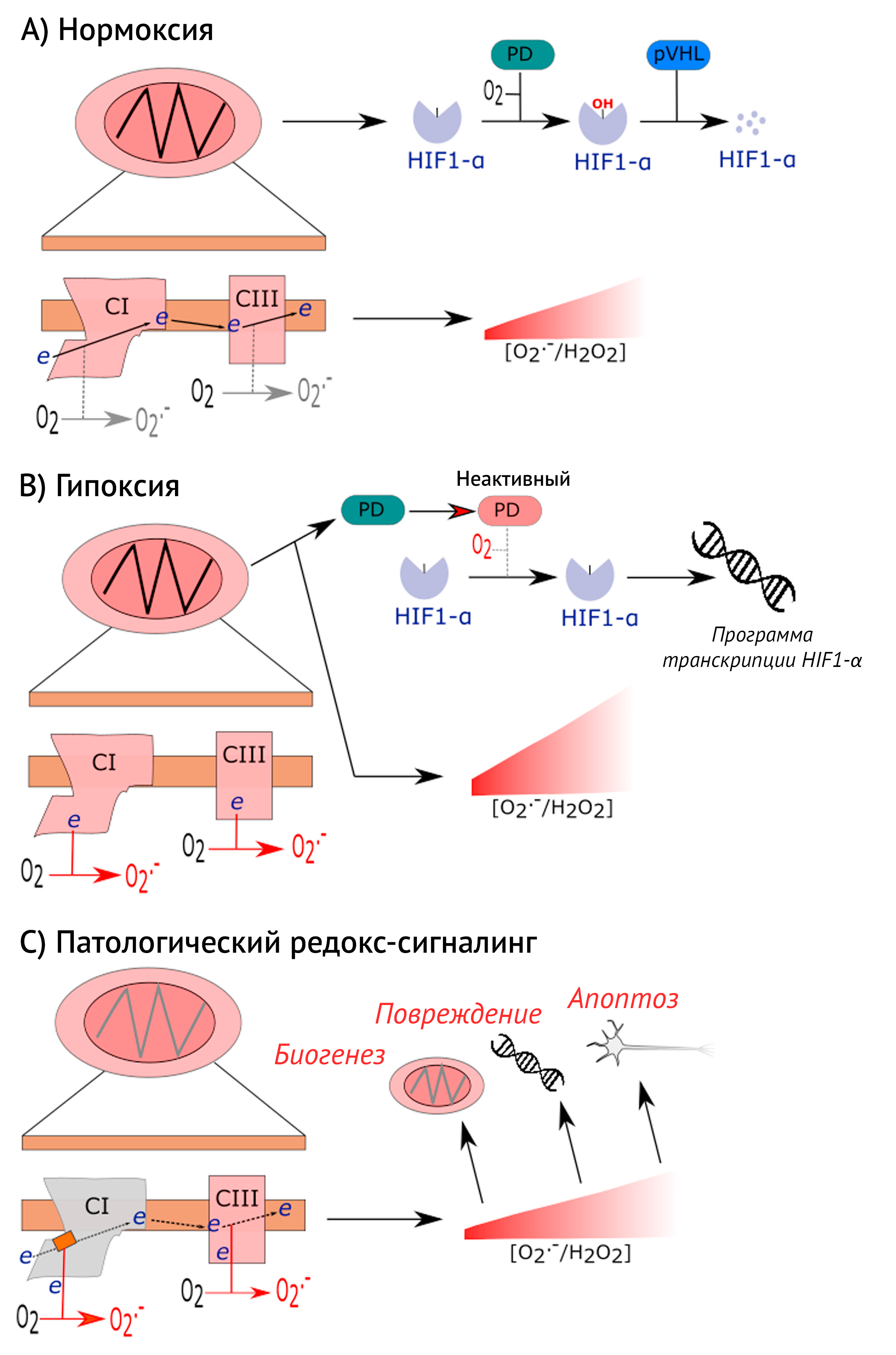
B) При гипоксии восстановленные комплексы I и III синтезируют O2•-/H2O2. O2•-/H2O2 инактивируют PHD, возможно, через высвобождение из активного сайта иона Fe2+. Ингибирование PHD позволяет HIF1-α попасть в ядро и запустить программы транскрипции генов, связанных с гипоксией.
C) Нарушение редокс-сигналинга. Серым цветом показан неправильно сложенный комплекс I (по причине мито-ядерного несоответствия днк). Неправильно собранный комплекс I перенаправляет электроны на O2 с образованием O2•-. В результате устойчивого синтеза O2•- в митохондриях могут происходить различные нарушения сигналинга, включая биогенез, повреждение ДНК и апоптоз.
Вместо того чтобы обсуждать прозаическую мысль о том, что O2• –/H2O2 являются обязательными токсичными побочными продуктами митохондриального дыхания, авторы рассматривают уязвимость нейронов к окислительному стрессу, вызванному митохондриями, с точки зрения сигналинга [111]. То, каким способом митохондрии синтезируют O2• –/H2O2 (для исчерпывающего разбора этого момента смотрите Мёрфи [68]), ставит их в поддержании нормальной функции органелл [112]. Целенаправленный синтез O2• –/H2O2 тесно связан с адаптивным редокс сигналингом [113].
Хорошим примером являются сигнальные пути, запускаемые при гипоксии. Митохондрии реагируют на гипоксию (то есть 0,3–3 % O2), синтезируя с помощью комплексов дыхательной цепи I и III O2• –/H2O2, которые активируют индуцируемый гипоксией фактор 1-альфа (HIF1-α), разрушая пролилгидроксилазу. HIF1-α запускает адаптивный ответ, инициируя транскрипцию ряда белков [114–118]. Так как продукция O2• –/H2O2 в митохондриях на отдельно взятом участке зависит от концентрации кислорода, числа восстановленных участков какого-либо из комплексов и кинетики реакции [68], гипоксия приводит к восстановлению комплексов I и III и запускает синтез O2• – (который может быть ускорен сниженной активностью COX и повышенной локальной доступностью O2). В ответ на клеточную гипоксию HIF1-α активирует транскрипцию NDUFA4L2, альтернативную субъединицу комплекса I, чтобы таким способом подавить синтез O2• – [119].
Нарушения редокс-сигналинга могут приводить к нейродегенерации:неспособность клетки остановить митохондриальную продукцию O2• – может запустить редокс-зависимый апоптоз по внутреннему пути [120,121]. Кроме того, рассогласованность дыхательной цепи вследствие нарушения взаимодействия митохондрий и ядра может запускать сигнал (т. е. O2• –/H2O2) без какого-либо стимула (т. е. гипоксии), приводя к неадекватному ответу [111]. Если возникает накопление мутантных митохондрий, они могут привести к дисфункции клетки путем клонального повышения своей численности, так как O2• –/H2O2 регулируют биогенез митохондрий [111,122,123].
6. Эндогенный метаболизм нейромедиаторов приводит к образованию пероксида водорода
Метаболизм эндогенных моноаминных нейромедиаторов (например, дофамина) приводит к образованию H2O2 при участии моноаминоксидаз. Моноаминоксидазы А и B катализируют реакцию дезаминирования: амин + O2 + H2O → альдегид + H2O2 + NH3. Обе изоформы фермента дезаминируют дофамин, тирамин, триптамин и норадреналин, МАО-А более избирательна к 5-гидрокситриптамину, МАО-B — к 2-фенилэтиламину [124,125]. В ходе катализа окисление амина до имина восстанавливает простетическую группу флавина, который затем вступает в реакцию с O2 с образованием H2O2 [126,127]. После восстановления флавина интенсивность синтеза H2O2 контролируется скоростью связывания O2, учитывая, что O2 влияет на ферментативную активность. Аффинность каждой из изоформ МАО к O2 составляет 10 и 240 µМ для МАО-А и МАО-B соответственно [127].
В условиях насыщения О2 их способность производить H2O2 оказывается внушительной: Cadenas и коллеги показали [128], что дезаминирование триптамина повышает уровень H2O2 в митохондриях мозга приблизительно на 1 нмоль/кг/мин. Очевидно, что присутствие фермента, синтезирующего H2O2, в потоке субстратов, зависимых от нейрональной активности, будет вызывать окислительный стресс [17,18]. В частности, считается, что МАО-B расположена на внешней стороне внутренней мембраны митохондрий [129], так как межмембранное пространство митохондрий (MIS) имеет способность частично нейтрализовать синтезируемый H2O2. Единственная пероксидаза MIS — глутатион пероксидаза 4 [59] — более избирательно восстанавливает гидропероксиды (ROOH), чем H2O2 [130,131]. Активность MАО-A/B может запускать апоптоз по Ca2+-зависимому пути [132–134], что позволяет проводить связь между синтезируемой моноаминооксидазами H2O2 и гибелью нейронов.
Неудивительно, что аномальная активность MАО-A/B связана со старением [135] и соответствующими нейродегенеративными заболеваниями, особенно БА и болезнь Паркинсона (БП) [136,137]. Это подогревает интерес к использованию синтетических ингибиторов MАО-A/B для лечения нейродегенеративных состояний и, разумеется, расстройств настроения [124,125,138]. Недооцененным аспектом работы MАО-A/B является то, что этот фермент ограничивает участие нейромедиаторов в O2• –-зависимом окислении и последующих циклах окисления-восстановления, в результате чего снижается синтез H2O2. Например, окисление дофамина может приводить к образованию нескольких молекул H2O2 [139], в то время как его окисление МАО-A/B приводит к образованию лишь одной молекулы пероксида с оговоркой, что определенные продукты реакции (например, 4-дигидроксифенилацетальдегид) могут участвовать в циклах окисления-восстановления [140,141]. Аналогичным следствием будет то, что ингибируя нейрональную активность изоформ МАО будет оказывать защиту против эксайтотоксичности. Кроме того МАО-А/B могут защищать мозг от экзогенных ксенобиотиков.
Несмотря на это, электрофильные альдегиды могут связываться с макромолекулами, вызывая их повреждение [142]. Например, 3,4-дигидроксифенилацетальдегид, производное дофамина, может быстро конъюгировать с белками и оказывать токсическое действие на нейроны [143], для гибели клетки достаточно повышения его концентрации с 2–3 до 6 мкм [144]. Поэтому активность изоформы МАО должна уравновешиваться активностью альдегиддегидрогеназы (АлДГ), чтобы предотвращать токсическое действие конечных продуктов. Но так как АлДГ-2 находится в матриксе митохондрий [145], MIS оказывается лишенным способности нейтрализовывать альдегиды ферментативным путем, это может способствовать формированию конъюгатов макромолекул, особенно если электрический заряд препятствует их пассивной диффузии. Возможно, в основе окислительного стресса, вызываемого МАО, лежат электрофильные альдегиды (в противоположность H2O2). Обсуждается, может ли H2O2 выступать в роли сигнальной молекулы, информирующей ядро о формировании альдегидов. Было показано, что ингибирование АлДГ способствует развитию БП [146] — это подчеркивает важность наличия активности, балансирующей работу МАО. Таким образом изоформы МАО могут вызывать окислительный стресс в головном мозге.
7. Аутоокисление нейромедиаторов
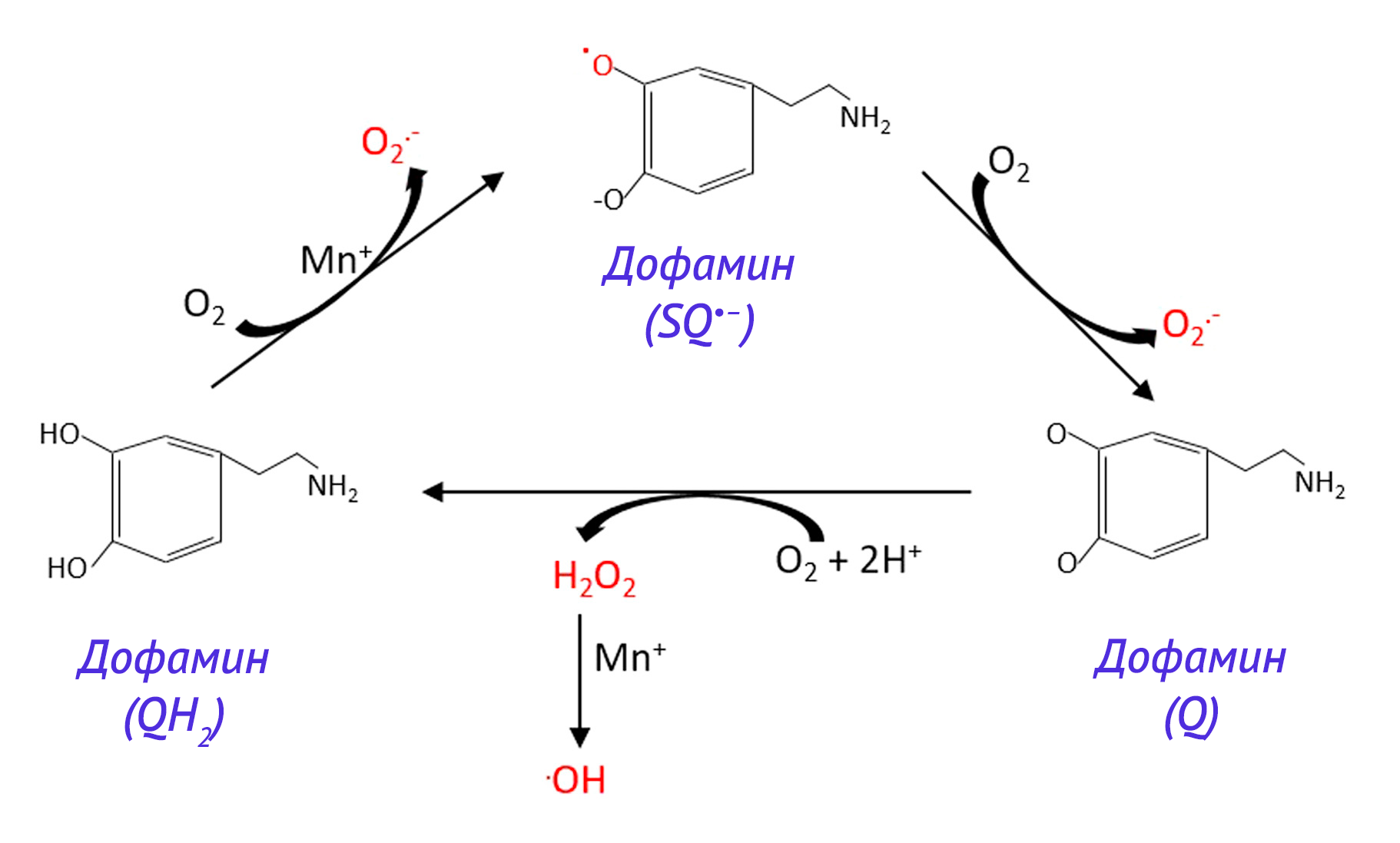
В своих основополагающих работах Кохен и Хейккиля [147,148] показали, что дофамин может взаимодействовать с O2 с образованием семихинонового радикала, который снова реагирует с O2, в результате чего образуется O2• – и хинон дофамина. Хотя в обычных условиях скорость синтеза семихиноновых радикалов невысока [19], эта реакция может быть катализирована ионами металлов с переходной валентностью [149], в изобилии присутствующих в головном мозге [17]. Хиноны дофамина могут реагировать между собой с образованием семихинонов, которые, в свою очередь, будут реагировать с O2 и формировать O2• –, однако стоит учесть, что эта реакция конкурирует с реакцией циклизации, тем самым препятствуя реакциям цикла окисления-восстановления [151]. Наличие смеси O2• –, H2O2 и •OH свидетельствует о равновесии гидрохинона, семихинона и хинона [150].
Продукты окисления дофамина могут участвовать в циклах окисления-восстановления [152]. Например, 6-гидроксидофамин может восстанавливаться до семихинон-радикала и H2O2. Часть H2O2 может реагировать с ионами металлов с переходной валентностью [11] и создавать условия для синтеза O2• – [139] и •OH. Повсеместно распространенные изоформы супероксиддисмутазы (СОД) [153–155] быстро нейтрализуют O2• – (k~2×109 M-1 с-1) и таким образом обрывают реакции образования радикалов [156]. Комплексное влияние изоформ СОД на O2• – достаточно сложно, так как они могут способствовать синтезу O2• – [139,150]. Снижая концентрацию O2• – вплоть до пикомолярных значений, изоформы СОД могут сдвигать равновесие реакций семихинонов в сторону их реагирования с O2 и образованием O2• –, как и в случае с 4-дигидроксифенилацетальдегид-радикалом [140].
Важно то, что способность к аутоокислению также имеется у серотонина и адреналина, более того, реакция аутоокисления адреналина используется для оценки активности СОД [157]. Поэтому нейромедиаторы с катехольной группой делают мозг особенно чувствительным к окислительному стрессу [17,18]. Например, циклы окисления-восстановления метаболитов дофамина, в частности, 6-гидроксидофамина, участвуют в развитии БП [158,159]. Окисление дофамина при БП [160] нарушает работу митохондрий и лизосом частично через формирование хинонов дофамина, изменяющих активность глюкоцереброзидазы — фермента лизосом, вовлеченного в патогенез БП [161], а также повышения концентрации H2O2 в митохондриях.
8. Скромные ресурсы антиоксидантной защиты
Как подробно рассматривается у Halliwell [17,18], относительно небольшие ресурсы антиоксидантной защиты повышают уязвимость мозга к окислительному стрессу. Другими словами, низкие способности эндогенной антиоксидантной защиты относительно других тканей (например, печени) делают мозг чувствительным к нарушениям редокс-гомеостаза. Известным примером является низкое содержание каталазы [163]: нейроны имеют в распоряжении в 50 раз меньшее количество фермента по сравнению с гепатоцитами [162], однако значение каталазы в инактивации H2O2 остается под вопросом.
Кроме того, что локализация каталазы ограничена пероксисомами, а механизм ее работы подразумевает реакцию одновременно с двумя молекулами H2O2 [164], что ограничивает активность фермента относительно наномолярных количеств H2O2 [164].
Более убедительным примером является глутатион. По сравнению с другими клетками цитозоль нейронов содержит вдвое меньшее количество глутатиона (например, ~ 5 мМ в нейронах против 10–11 мМ в гепатоцитах). Низкое содержание глутатиона, в свою очередь, является следствием ограниченных способностей нейронов его синтеза ввиду малых количеств γ-GCL (γ-глутамилцистеинлигазы), причиной чего, в свою очередь, является низкие содержание и активность Nrf-2 [81,166]. Сравнительно малые ресурсы цитозольного глутатиона могут ограничивать активность GPX4 [130], что может объяснять повышенную уязвимость нейронов к ферроптозу [167]. Малые количества глутатиона могут также ограничивать способность нейронов метаболизировать электрофилы, в частности электрофильные альдегиды.
Из того, что уже обсуждалось выше, может показаться, что данный дефект связан с метаболизмом H2O2 [18,19] ввиду стандартного содержания и активности изоформ СОД (то есть при отсутствии нарушений в метаболизме O2• –). Поддержание нормальной инактивации O2• – (например, при помощи СОД) жизненно необходимо для нейронов: при делеции гена MnSOD (митохондриальной изоформы СОД) нервные клетки погибают [168–170]. В итоге ферментные системы нейронов, связанные с глутатионом, являются недостаточными.
В 1994 году группа под руководством Су Гу Ри обнаружили семейство ферментов PRDX — повсеместно распространенных цистеин-зависимых пероксидаз [171]. Изоформы PRDX восстанавливаются ферментом TRDX (тиоредоксин), а окисленный TRDX, в свою очередь, восстанавливается тиоредоксин-редуктазой за счет НАДФH [172–176]. Это открытие оказало важное влияние на понимание того, как нейроны нейтрализуют H2O2, так как нервные клетки экспрессируют изоформы PRDX-TRDX [162,177]. В норме система PRDX-TRDX позволяет нейронам нейтрализовывать H2O2, особенно если учесть что изоформы PRDX обильно распространены и распределены внутри клеток [171,175,178]. Авторы не знают ни одного отчета о слабой активности нейрональной системы PRDX-TRDX. Напротив, нейроны могут обладать относительно высокой активностью PRDX, так как метаболизм глюкозы нервных клеток идет преимущественно через ПФП, в котором генерируется НАДФH [95]. Однако есть ферменты, которые могут использовать НАДФH для синтеза O2• – и NO• (например, NOS) [179,180], поэтому будет опрометчиво считать, что НАДФH находится исключительно на стороне «антиоксидантов» [181]. Тем не менее, PRDX6 в качестве восстановителя предпочитает глутатион [182], поэтому относительно низкое содержание последнего может ограничивать активность и этого фермента. Не стоит считать, что система PRDX-TRDX занимается только нейтрализацией H2O2, так как есть убедительные биохимические доказательства участия PRDX-TRDX в передаче редокс-сигналов [16,172,183–187].
Как элегантно сформулировали Флоэ с соавт. [188], вряд ли природе понадобилось бы создавать 10 пероксидаз, чтобы просто обезвреживать H2O2. Лу, Хольмгрен с соавт. [162] предположили, что наличие PRDX-TRDX дает нейронам способность обуздать их относительную склонность к окислительному стрессу и направить его на передачу редокс-сигналов. Если это так, то подобное положение дел опасно, потому что при увеличении содержания H2O2 до высоких наномолярных концентраций будет происходить переокисление изоформ PRDX, что является признаком гибели клетки [45,180]. Это становится особенно актуальным, если учесть слабую активность глутатион-зависимых ферментных систем нейронов [81,166]. Тандем PRDX-TRDX дает возможность как нейтрализовывать H2O2, так и направлять H2O2 на потребности клеточного сигналинга, однако это остается возможным до тех пор, пока концентрация H2O2 остается ниже критического порога, после которого уже развивается окислительный стресс.
9. Микроглия
Микроглия представлена специализированными резидентными иммунными клетками [189,190], постоянно «патрулирующими» свою нишу на предмет угроз для гомеостаза [191,192]. Микроглиоциты образуют обширные отростки для оценки состояния синапсов и отслеживания активности нейронов [193]. Осуществляя мониторинг нейрональной активности, микроглия выполняет важную функцию по элиминации нездоровых нейронов, формировании межнейронных связей на стадиях развития организма и синаптической пластичности, зависящей от активности синапсов [194–197].
Прорывная фундаментальная работа Бернарда Бабиора [198] показала, что активные иммунные клетки синтезируют O2• – при помощи изоформ NOX (преимущественно NOX2 [23]). Использование O2• – для уничтожения бактерий было одним из первых примеров биологически полезных функций, открытых у свободных радикалов [199]. Поэтому неудивительно, что микроглия во время фагоцитоза синтезирует O2• – и родственные ему соединения [200]. Однако стоит иметь в виду, что продукция O2• – зависит от наличия O2, и поэтому активность микроглии крайне чувствительна к местной доступности кислорода для тканей: использование ими кислорода для синтеза O2• – вместо АТФ может быть даже одним из способов элиминации синапсов. Маловероятно, что малоактивные (например .OH) или анионные (например O2• –) АФК были способны покидать фагосомы и повреждать соседние нейроны (если анионы, все-таки, покидают фагосомы, их вход в нервные клетки в любом случае ограничивался бы их зарядом). Не несущие заряда H2O2 и NO• могут легко диффундировать сквозь мембраны и вызывать повреждение или усиление местного воспаления путем привлечения большего числа микроглии.
Группы под руководством Niethammer и Amaya показали, что при заживлении ран и регенерации конечностей H2O2 действует как хемоаттрактант [201,202]. Глия, патрулирующая свою территорию, может «чувствовать» H2O2 и отвечать на это активацией и пролиферацией [203], что объясняет, почему высвобождение H2O2 из нейронов привлекает микроглию.
Кроме того, тирозин-киназа Lyn детектирует нейрональную H2O2 и запускает хемотаксис микроглии через F-актин [204]. Каким образом Lyn обнаруживает H2O2, остается неясным, однако этот механизм может включать инактивацию H2O2-связанной фосфатазы [15,16]. Процесс воспаления может усиливаться благодаря цитокинам и активности микроглии, поддерживающей перекисное окисление липидов (ПОЛ) [205,206]. Хотя микроглия необходима для нормального функционирования и развития мозга, нарушение ее активности может провоцировать окислительный стресс [206]. Например, при БА микроглия способствует нейродегенерации, избыточно активируя прунинг синапсов [207,208]. Требует ли избыточный прунинг запуска окислительного стресса — неизвестно. Возможно, метаболиты ПОЛ (например, 4-гидроксиноненаль) [209] привлекают микроглию путем модификации цистеиновых остатков белков через реакцию Михаэля [210,211].
10. Металлы с переходной валентностью
Редокс-активные металлы с переходной валентностью (такие как Fe2+ и Cu+) присутствуют в головном мозге в значительных количествах [17,18]. Относительный избыток подобных металлов выражается в том, что их концентрация в 10000 раз превышает содержание нейромедиаторов [212,213]. Одна только концентрация связанного Fe2+ может достигать уровня миллимолей. Большой биохимический потенциал Fe2+ и Cu+ используется для катализирования химических реакций. Например, Fe2+ может связываться с электронно-плотными группами O и N органических молекул [214]. Соответственно, ионы Fe2+ (и Fe3+) необходимы для обеспечения каталитической активности ряда ферментов, например: аконитазы, фумаразы и цитохромов P450 [214]. Кроме того, Fe2+ принимают участие в синтезе миелина [215,216] в качестве кофактора ферментов, осуществляющих синтез липидов de novo [215,216]. Нейроны также содержат малосвязанный пул Fe, называемый лабильным пулом железа (ЛПЖ), который поддерживается поступлением с пищей и в большинстве тканей составляет около 20 мкм [167,217].
Относительный избыток железа означает, что нейроны должны строго контролировать уровень O2• –/ H2O2, чтобы избежать риска неправильной металлизации — следствия реакции замещения с образованием FeS [218] — и реакции Фентона, которая в качестве побочного продукта производит .OH (Fe2+ + H2O2 → OH– + .OH) [19]. Даже с учетом большого содержания и быстрой кинетики СОД, поддерживающих постоянную концентрацию O2• – на пикомолярном уровне, по оценкам Imlay [219], период полуповреждения мономолекулярного фермента по-прежнему составляет ~ 20 мин ввиду хорошей кинетики реакции (k ~ 106 М-1 с-1). Например, потеря иона Fe2+ приводит к инактивации рибулозо-5-фосфат 3-эпимеразы, фермента ПФП [220]. Такая инактивация может быть обратимой при наличии компенсаторного механизма (например, включения иона Mg2+, или LIP-опосредованного замещения ионом металла [220]). Несмотря на это, нарушение работы ПФП из-за инактивации рибулозо-5-фосфат 3-эпимеразы может негативно сказываться на работе нейронов по причине снижения синтеза нуклеотидов [221].
Особенно интересно то, что Fe2+ управляет реализацией ферроптоза — относительно недавно открытой формы клеточной смерти, зависимой от Fe2+ и ПОЛ [88,167]. Fe2+ запускает ферроптоз, катализируя реакцию образования пероксил- и алоксил-радикалов из ROOH. Кинетически эти реакции протекают легче (k ~ 1,3 × 103 M-1 s-1), чем реакция Фентона (k ~ 76 M-1 с-1) [19,222,223]. Однако роль Fe2+ достаточно сложна, так как Fe2+ могут и ингибировать ПОЛ путем связывания ROO. и RO. в более быстрых реакциях (например RO. + H+ + Fe2+ → ROH + Fe3+, k ~ 3,0 × 108 M-1 с-1) [222]. Но низкое отношение концентраций ROO./RO. означает, что ионы Fe2+ с большей вероятностью прореагируют с ROOH. Ввиду отношения этих реакций мозг оказывается уязвим к нарушению гомеостаза железа [224]. Такая повышенная чувствительность мозга к нарушениям метаболизма Fe2+ подчеркивается наблюдениями, что комплексы ионов железа с бета-амилоидом способствуют отложению бляшек при БА [225].
Аналогично ионам Fe2+, нейроны содержат лабильный пул Cu+ [226] который, кажется, также важен для передачи сигналов и процессов возбуждения в нервных клетках [227,228]. Например, нейроны могут управлять своей спонтанной активностью, перераспределяя слабо связанный пул Cu+ из сомы в дендриты [227,229,230]. Кроме того, Cu+ является необходимым кофактором ферментов [212], среди которых ярким примером является ЦОГ и медь-цинк-СОД (Cu,Zn-СОД) [153,231,232]. Высокое содержание Cu+ в нейронах (от 0,1 мм до 1,3 мм в определенных областях) создает условия, в которых Cu2+ будет катализировать реакцию Фентона и окисление белков образовавшейся H2O2 [212]. Потенциальные рамки, в которых может нарушаться гомеостаз Cu+ можно хорошо проиллюстрировать на примере спорадического БАС. В частности, мутантные варианты Cu,Zn-СОД способствуют развитию как семейной, так и спорадической формам БАС [233]. Каким именно образом нарушение работы Cu,Zn-СОД способствует развитию БАС, до конца не понятно, однако оно может быть связано с накоплением токсических агрегатов белков, утративших свои функции, активностью пероксидазы, которая может генерировать CO3- через HOOCO2 [231,236] и активностью тиолоксидазы [237,238]. Кроме того, снижение активности Cu,Zn-СОД будет приводить к повышению концентрации ONOO- [239].
11. Высокое содержание ПНЖК
Головной мозг является большим депо омега-3 ПНЖК (полиненасыщенных жирных кислот) [240], особенно докозагексаеновой кислоты (ДГК). Учитывая энергетические потребности мозга, можно ожидать, что нейроны будут заниматься окислением жирных кислот для получения АТФ, особенное учитывая его большие концентрации: 106 АТФ на одну молекулу пальмитиновой кислоты, 32 АТФ на одну молекулу глюкозы [241], и только 14–17 АТФ на молекулу лактата [3]. Однако скорость бета-окисления в мозге существенно ниже [241] по сравнению с другими метаболически активными тканями (например, скелетной мускулатурой [242]). Возможно, в свете низкой активности каталазы [163] становится выгодным ограничивать использование кислорода на окисление пальмитиновой кислоты (ее окисление требует на 15 % больше O2), и таким образом снизить синтез H2O2 в пероксисомах [243,244]. Подобный отказ от окисления липидов для получения АТФ может быть обусловлен использованием липидных пероксидов в качестве сигнальных молекул [19]. Например, ДГК может быть превращена в резольвины, обладающие противовоспалительными эффектами [245]. Кроме этого, жирные кислоты и холестерин востребованы для синтеза миелина. Важность последнего для мозга подчеркивается тем, что он содержит около 20 % всего холестерина организма [19].
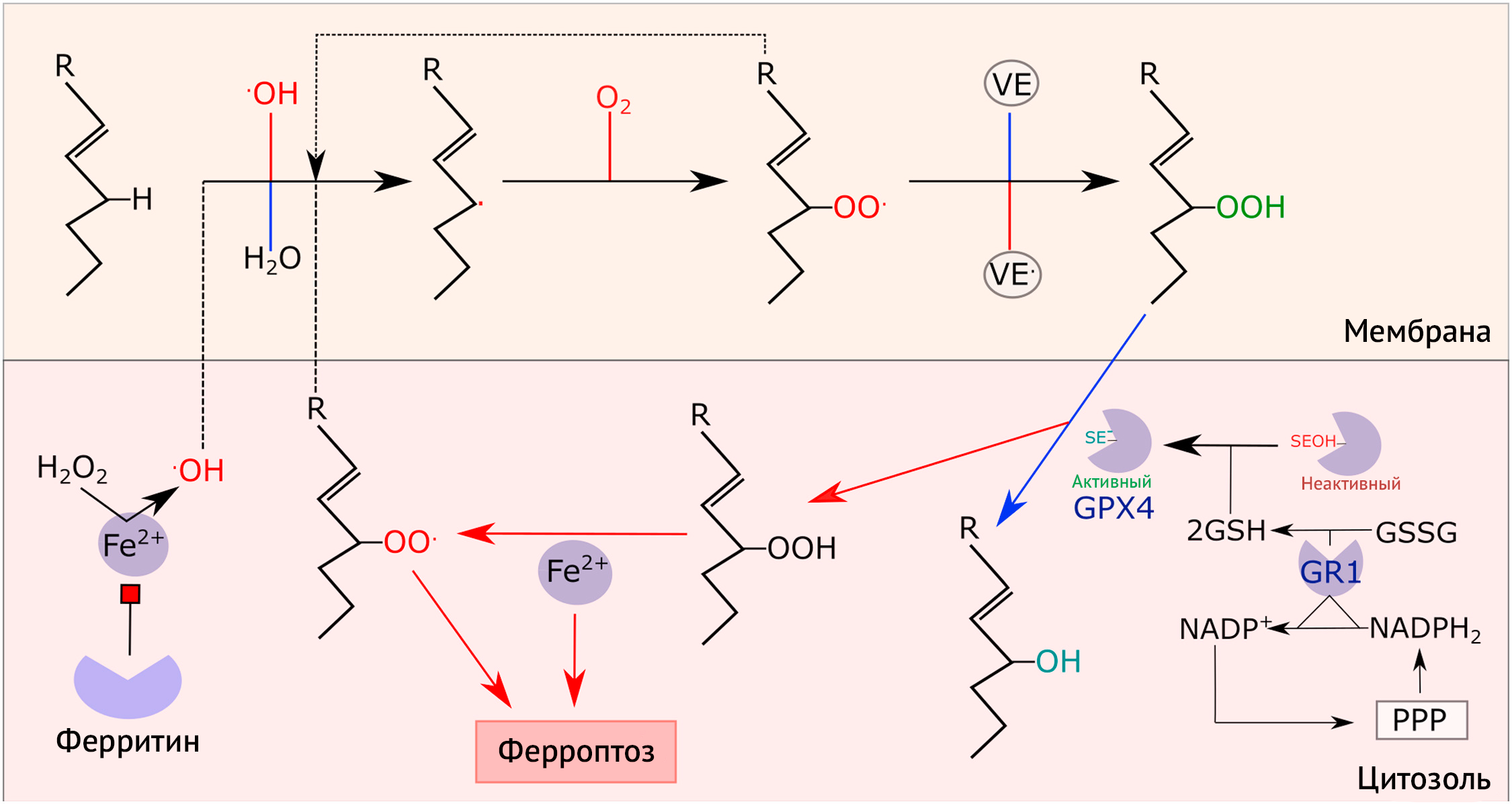
2) Развитие цепи (оксигенация). Синтез ROO. в результате реакции углеродного радикала с O2
3) Разрушение структуры липидов (амплификация). ROO. отщепляет H+ от ненасыщенных липидов с образованием ROOH и углеродного радикала
4) Обрыв цепи (терминация). Альфа-токоферол (витамин Е) обрывает цепи радикалов, вступая в реакцию с ROO. и образуя ROOH.
5) GPX4. Глутатионпероксидаза 4 превращает ROOH в соответствующий спирт. Сама GPX4 затем восстанавливается двумя молекулами глутатиона. Образовавшийся GSSG, в свою очередь, восстанавливается глутатионредуктазой, использующей НАДФH, полученный из ПФП.
6) Ионы железа. Fe2+ реагирует с ROOН с появлением ROO., который может инициировать ПОЛ и при условии сниженной активности GPX4 запускать ферроптоз. Кроме того, при участии Fe2+ H2O2 может превращаться в .OH (реакция Фентона), что может снова инициировать ПОЛ. Ферритин может сдерживать ПОЛ, связывая ионы Fe2+.
Для наглядности были опущены некоторые этапы (например, перестройки углеродных радикалов).
Холестерин может аутоокисляться свободными радикалами или же через нерадикальные механизмы [246]. Высокое содержание липидов с ПНЖК определяет высокий риск развития окислительного стресса, так как ПНЖК очень чувствительны к ПОЛ — это может оказаться биологической расплатой за использование липидных пероксидов в качестве сигнальных молекул [89]. Перекисное окисление липидов (подробнее рассмотрено в [43,247–250]) включает начальную генерацию углеродного радикала, после чего может следовать реакция присоединения или отщепления при участии АФК достаточной активности (например, •OH) по метиленовой группе. К слову, наличие •OH может быть необязательным для инициации процессов ПОЛ, в качестве запускающего фактора могут выступать гипервалентные формы железа (например FeIV=O) [222].
Углеродные радикалы также могут реагировать с кислородом с образованием ROO•, который может забирать H+ в бис-аллильных положениях других молекул и таким образом продолжать цепь реакций с образованием ROOH и углеродного радикала [43,247–250]. Альфа-токоферол (α-TOC) может вмешиваться в такие цепи реакций и прерывать их, превращаясь в резонансно стабильный радикал [249]. Однако ROOH может реагировать с металлами с переходной валентностью и снова образовывать ROO• и RO• [222]. GPX4 способна нейтрализовывать ROOH до ROH [251], но далее необходима реактивация фермента при участии GSH, реактивация которого в свою очередь требует работы НАДФH-зависимой глутатион редуктазы [130,131,188].
Такой феномен как ферроптоз [90, 252] объясняет, почему делеция по гену GPX4 приводит к гибели эмбрионов [253,254], так как GPX4, нейтрализуя ROOH, выступает в роли важного регулятора ферроптоза [223,255] — наличие и отсутствие активности GPX будет, соответственно, останавливать или запускать ферроптоз [256]. Низкие концентрации GSH повышают уязвимость нейронов к ферроптозу, что подтверждается гибелью нейронов при условном «выключении» GPX4 [257–259]. Это согласуется с вкладом ПОЛ в патогенез нейродегенеративных заболеваний (например, БА [209,211]).
12. Мозг использует NOS и NOX для передачи сигналов
При выполнении своих ключевых функций мозг полагается на изоформы nNOS и NOX. В свою очередь, синтез NO• при участии nNOS требует наличия O2, НАДФH и L-аргинина [37]. Аффинитет nNOS к O2 составляет 300 µM, это значит, что при средней концентрации O2 в мозге около 20 µM синтез NO• будет значительно ограничен [260]. Однако NO• регулирует такие важные физиологические процессы, как ДВП (долговременная потенциация) [261,262], рост аксонов [263] и прунинг [264]. Как уже было сказано, NO• может провоцировать развитие окислительного стресса — особенно когда одновременно происходит синтез O2•-. Биохимия nNOS подразумевает пространственно близкий синтез как NO•, так и O2•-, что повышает вероятность образования ONOO– [265]. При работе несвязанной nNOS эти реакции проходят параллельно. Рост количества свободной nNOS наблюдается, когда окисляются или отсоединяются важные кофакторы (например, тетрагидробиоптерин).
Вторая группа ферментов, изоформы NOX, используют простетические редокс группы для окисления НАДФH и восстановления O2 до O2•- [22,23]. Наличие изоформ NOX важно для мозга (подробнее в [266]), например для ДВП и работы микроглии [24].
Так как синтез O2•-, опосредованный изоформой NOX, далеко не случайно регулируется на нескольких уровнях [22,23,267], связанный с NOX окислительный стресс обычно возникает под действием длительных и нежелательных сигналов, идущих вместе с постоянной подставкой НАДФH и O2, поддерживающих активность фермента. Такая ситуация может происходить при развитии нейровоспаления [206], когда цитокины запускают и поддерживают синтез O2•- при участии NOX2 микроглии [205].
13. Окисление РНК
Окисление РНК редко рассматривается как причина повышенной чувствительности мозга к окислительному стрессу [268]. Кроме основной матричной РНК мозг в значительной мере полагается на некодирующую РНК, особенно на длинные некодирующие РНК и микроРНК (подробнее в [269–271]). С биохимической точки зрения РНК также уязвима для окисления и подвергается аналогичным реакциям, что и ДНК [272]. Например, 8-оксогуанин является результатом окисления как РНК, так и ДНК [273]. Но ввиду своей одноцепочечной структуры РНК уязвима не только к окислению, но и к алкилированию [274] по местам образования водородных связей. Кроме того, РНК лишена защитного эффекта гистонов, а также не компарментализована (ДНК находится за ядерной мембраной), например, при нахождении в аксонах и синапсах. Хотя для функционирования синапсов очень важен локальный синтез белка [275,276], пока не изучено, будет ли окисление РНК как-то нарушать этот процесс.
Окисление РНК приводит к появлению ошибок в ее коде, что, в свою очередь, останавливает синтез белка на рибосомах [277], и при отсутствии вмешательства систем репарации могут образовываться неполные, мутантные или неправильно сложенные белки [278]. Близкое расположение мРНК к митохондриям и временная динамика окисления РНК (секунды) по сравнению с с трансляцией (часы) делают окисление РНК высоко вероятным, особенно в нейронах, где присутствуют металлы с переходной валентностью, способные участвовать в реакции Фентона [279–281].
Подход в рассмотрении окисления мРНК как причины окислительного стресса, связанного с нейродегенерацией, подтвеждается наблюдением, что окисленная мРНК Cu,Zn-СОД является ранним доклиническим признаком БАС [282].
Согласно ряду отличных обзоров [268,272], необходима дальнейшая работа для понимания распознавания, оборота и восстановления окисленной РНК [283]. Только с лучшим пониманием каждого из этих процессов можно будет оценить чувствительность нейронов, так как концентрация окисленной РНК является функцией от процессов ее образования и элиминации по времени.
Перспективы
Авторы хотят предложить всеохватывающий подход для выяснения причин, почему редокс-сигналинг приводит к развитию окислительного стресса в головном мозге. Конечной расплатой за использование редокс-сигналинга является неотъемлемая уязвимость головного мозга к окислительному стрессу в случаях, когда сигналы идут не так, как им следует, что, кажется, как раз и происходит при заболеваниях. Некоторые нейрофизиологи могли бы отрицать центральное значение нейрональной активности в этом вопросе. Но учитывая то, как митохондрия синтезирует O2•-/H2O2 [68], активность нейронов должна опосредованно регулировать митохондриальный синтез O2•-/H2O2. В активном синапсе потребности АТФ, если они могут быть обеспечены, должны снижать суммарный синтез O2•-/H2O2 в митохондриях. В это же время в неактивном синапсе низкие потребности в АТФ и ослабленная работы цепи переноса электронов должны благоприятствовать синтезу O2•-/H2O2, что потенциально ставит митохондриальные O2•-/H2O2 на роль своеобразных часовых синаптической активности. Если это так, то можно объяснить, каким образом высвобождение митохондриального O2•-/H2O2 в условиях неактивности синапса запускает долговременную депотенциацию (ДВДП) и даже синаптический прунинг — особенно, если постоянно используется один и тот же путь передачи сигнала [284].
Митохондриальный апоптоз управляет ДВДП и прунингом [285–287]. Активность митохондрий, связанная с высвобождением O2•-/H2O2, может провоцировать локальный сублетальный внутренний апоптоз и вызывать ДВДП и прунинг синапсов. Возможно, такой редокс-управляемый апоптоз является механизмом регуляции прунинга в развивающемся мозге — важного этапа, предшествующего формированию сложного коннектома, а также обязательного условия для коррекции нервных связей во взрослом возрасте [284]. Расположение митохондрий в синаптических окончаниях повышает риск потери синапсов. В случаях, когда митохондрии не могут обеспечить потребности в АТФ или же в условиях дефицита кислорода происходит высвобождение O2•-/H2O2, которые могут воспроизводить сигнал прунинга и вызывать нежелательное сокращение числа синапсов.
Имеется биологический прецедент: нежелательная реактивация сигналов прунинга, которые в норме действуют на этапах развития, способствует утрате синапсов при БА [207]. В биохимически оправданных 13 причинах уязвимости мозга к окислительному стрессу авторы умышленно приняли глобальную точку зрения, фокусируясь на «нейронах» как коллективе, работающем над общей задачей. Существуют параллели между общепринятыми терминами нейронов и активных форм кислорода [20]. Активные формы подразумевает химически гетерогенную совокупность соединений, которые могут на порядки различаться по скорости реакций с отдельно взятым субстратом (например, с гуанином •OH реагирует по мере диффузии, в то время как O2•- оставляет гуанин нетронутым ввиду низкой реактивности).
Аналогично активным формам кислорода, нейроны имеют гетерогенную природу, которая не укладывается в широкие определения, так как они могут сильно различаться по множеству ключевых параметров, включая функцию, расположение, связи, миелинизацию и длину аксона. Разнообразие нейронов определяет и различную чувствительность к окислительному стрессу как в пределах нейрона (т. е. тело или синапс), компартмента (т. е. синаптическая митохондрия или синаптическая мембрана), так и между нейрональным популяциями.
Дофаминергические нейроны pars compacta черной субстанции имеют разную чувствительность: они испытывают фоновый (то есть без дополнительных колебаний гомеостаза) окислительный стресс, так как их автономная пейсмекерная активность контролируется кальциевыми каналами L-типа, определяющими митохондриальный уровень O2•-/H2O2 [288]. При таком балансировании на грани окислительного стресса даже малейший неожиданный сдвиг во внутриклеточной редокс среде, возможно, связанный с метаболизмом дофамина [160], будет достаточным для того, чтобы привести к гибели.
Чувствительность нейронов к окислительному стрессу колеблется. В то время как постоянная концентрация O2•- отражает динамику скорости его синтеза и элиминации в отдельно взятом компартменте [68], чувствительность нейронов к окислительному стрессу на протяжении времени будут определять множество взаимосвязанных факторов. В качестве известного примера инициации ферроптоза авторы кратко рассмотрели восстановление ROOH до ROO. при участии Fe2+ [223].
Биомолекулярная реакция второго порядка характеризуется постоянной скоростью, средней концентрацией ROOH и Fe2+. Возможность синтеза ROO• зависит от доступности субстрата в данный момент времени: хорошими примерами являются утилизация ROOH при помощи GPX4 и связывание Fe2+ ферритином. Если связывание ксенобиотиков с GSH снижает его локальную концентрацию и таким образом ослабляет активность GPX4, это может способствовать синтезу ROO•.
«Прошлое» нейрона также влияет на его чувствительность к редокс-стрессу, и это усложняет ситуацию. Например, синаптическая активность, связанная сублетальными изменииями в редокс-статусе, определяет адаптивную координированную нейроглиальную реакцию, способствующую повышению внутриклеточного содержания GSH (подробнее в [81]).
В нашем примере адаптированный нейрон способен лучше нейтрализовывать ксенобиотики и препятствовать росту содержания ROO• и окисленных липидов, необходимых для осуществления ферроптоза [89]. Для предостережения стоит сказать, что адаптация требует частой стимуляции, так как концентрация GSH зависит от транскрипции на многих уровнях. В качестве интересной параллели можно привести аксиому физиологии упражнений «используй или потеряй»: при постоянной активности возникает комплекс адаптивных реакций к редокс-стрессу, однако при неактивности сопротивляемость прогрессивно снижается.
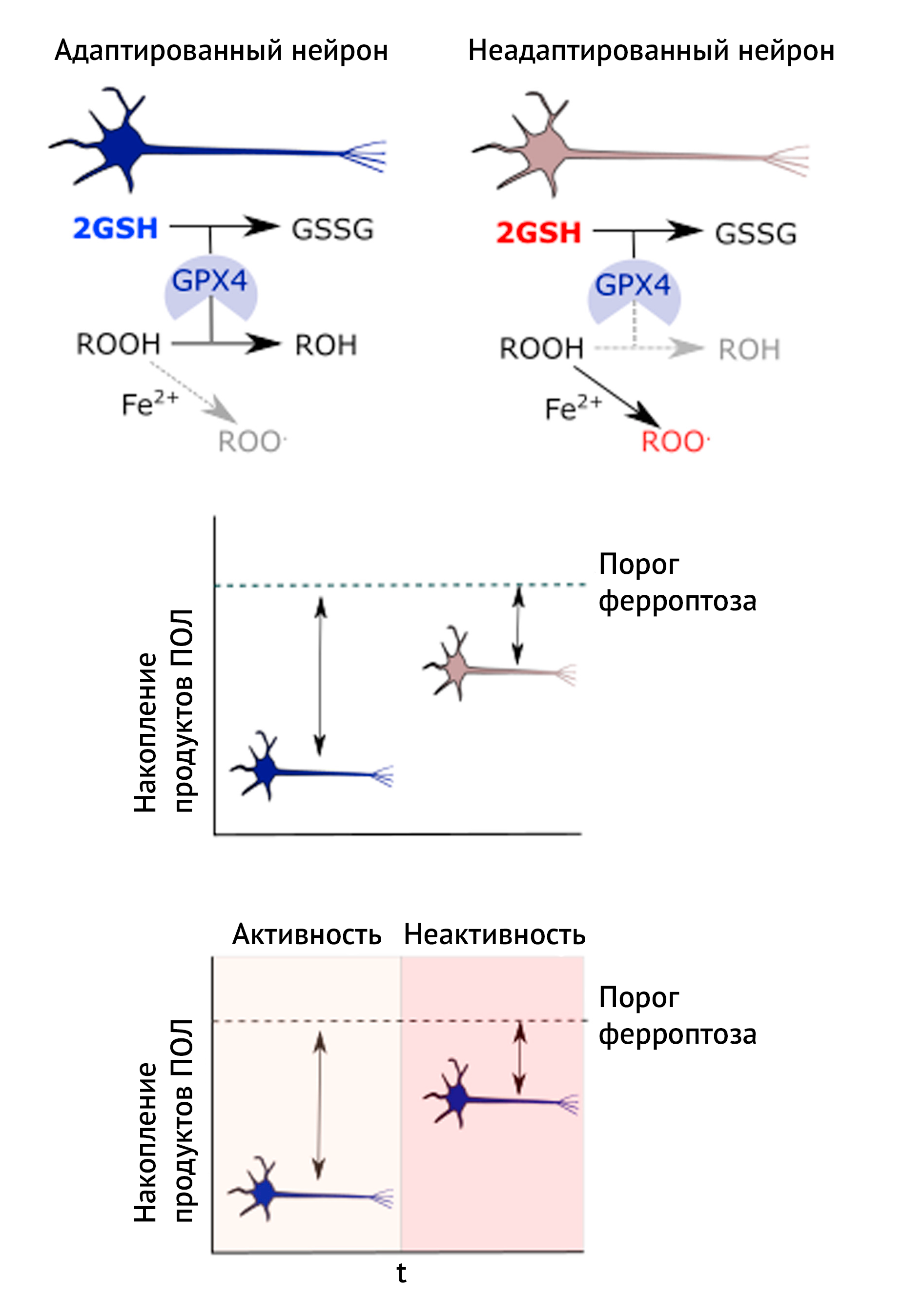
2) Теоретическая модель порога, когда предшествующая адаптация (которая описана выше) определяет содержание липидных пероксидов, требуемых для инициации ферроптоза. Адаптированный нейроне может выдержать больший подъем концентрации ROOH, прежде чем погибнуть ферроптозом.
3) Теоретическая пороговая модель, в которой адаптированный нейрон теряет устойчивость к ферроптозу в результате отсутствия активности. Таким образом, отсутствие стимулов (то есть нейрональной активности) нарушает адаптацию.
С трансляционной (трансляции в смысле переноса опыта доклинических исследований в клинические — прим. перев.) точки зрения высокая сложность редокс-гомеостаза нервных клеток помогает объяснять неуспешность попыток использования пищевых антиоксидантов для лечения нейродегенеративных заболеваний [289]. Кроме проблемы биодоступности, их неудача связана еще с кинетическими и пространственными ограничениями (подробнее в [181,290–293]). Вероятность существования соединения, обладающего достаточной биохимической универсальностью, чтобы одновременно и значительно влиять на каждый из обсуждаемых факторов, крайне мала. Прежде всего, неудачи пищевых антиоксидантов неотъемлемо связаны с их биохимическими ограничениями, однако этого недостаточно для отрицания причинной роли окислительного стресса. Многое в редокс-гомеостазе в норме и патологии головного мозга остается непонятным. Рациональная терапия нейродегенеративных заболеваний, направленная на процессы окисления-восстановления, может быть разработана только тогда, когда фундаментальные исследования раскроют новые детали механизмов регуляции редокс-гомеостаза.
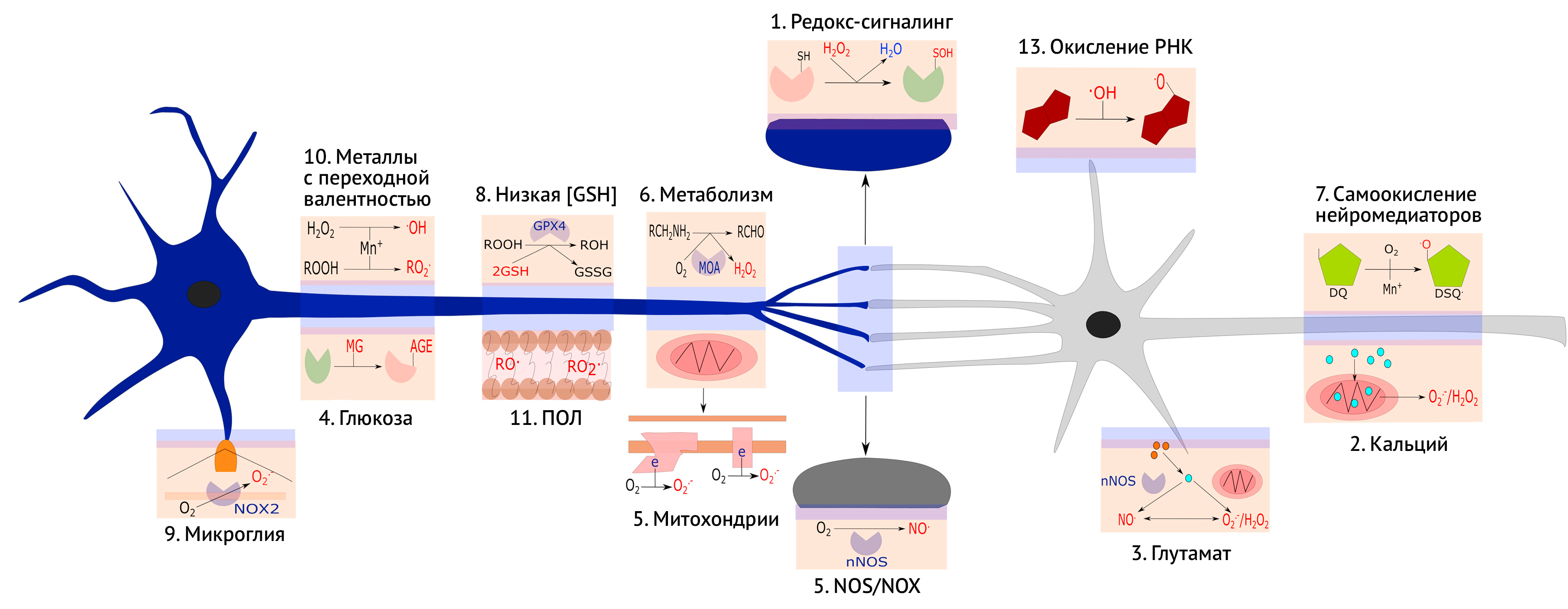
2) Кальций. Перегрузка митохондрий ионами Ca2+ вызывает синтез O2•-/H2O2.
3) Глутамат. Глутамат вызывает высвобождение ионов кальция, повышающих синтез NO. через активацию nNOS и O2•-/H2O2 в митохондриях, в результате чего образуется ONOO-, оказывающий нейротоксическое действие.
4) Глюкоза. Показана инактивация белков через образование AGE.
5) Митохондрии. Показан синтез O2•- комплексами I и III.
6) Метаболизм. Изоформы МАО катализируют синтез H2O2.
7) Окисление нейромедиаторов. Металлы с переходной валентностью (Mn+) катализируют аутоокисление дофамина до полухинон-радикала.
8) Слабая антиоксидантная защита. Низкая активность GPX4 связана с недостаточным количеством GSH.
9) Микроглия. Показан NOX2-опосредованный синтез O2•- в области окончания отростка нейрона.
10) Редокс-активные металлы с переходной валентностью. Показаны как Mn+, катализируют синтез ROO. и .OH.
11) Перекисное окисление липидов. Образование RO. и ROO. в цитоплазматической мембране нейронов.
12) Экспрессия NOS/NOX. Эти ферменты могут синтезировать NO..
13) Окисление РНК. В качестве примера показано окисление гуанина в РНК при участии .OH.
Заключение
Огромное число сложно взаимосвязанных факторов делают мозг уязвимым к окислительному стрессу, к первым 13 (существует гораздо больше [17,18]) относятся высокое содержание ПНЖК, глюкоза, митохондрии, кальций, глутамат, слабые системы антиоксидантной защиты, металлы с переходной валентностью, аутоокисление нейромедиаторов и РНК. Мозг чувствителен к окислительному стрессу также ввиду использования широкого спектра химически активных соединений для реализации большого арсенала сигнальных функций, начиная с липидных радикалов, запускающих ферроптоз при нарушении липидного сигналинга и заканчивая NO•, тонко управляющим синаптической пластичностью и митохондриальными O2•-/H2O2, передающими сигналы о гипоксии. Уже сам по себе баланс между специфическими биохимически полезными и вредными соединениями прекрасен, но подобная ситуация равноценна игре с огнем: при неверном потоке электронов сложный редокс-сигналинг легко может развиваться до окислительного стресса.
