
COVID-19, старение и нейродегенерация

SARS-CoV-2 — седьмой коронавирус, патогенный для человека [6]. Он относится к семейству Coronaviridae, группе больших оболочечных РНК-содержащих вирусов с положительной цепью. Это семейство включает в себя как менее патогенные вирусы, например HKU и 229E [7], так и высокопатогенные, ставшие причиной многих человеческих жертв, SARS-CoV и MERS-CoV, выявленные в 2002 и 2012 гг., соответственно [8]. Хотя сразу после выявления SARS-CoV-2 его начали сравнивать с H1N1 вирусом гриппа (ВГ), молекулярная структура этих вирусов значительно отличается.
Однако, наблюдаются некоторые сходства в том, как H1N1 и SARS-CoV активируют иммунную систему. Оба вируса изменяют работу эпигенетических механизмов, обусловливая избыточную активацию генов, стимулируемых интерфероном, которые служат первой линией обороны против вирусов [9].
SARS-CoV-2 лишь недавно оказался в поле зрения ученых, и корпус знаний о нем растет с каждым днем. В настоящее время еще не существует этиологического лечения или вакцины. На данный момент долгосрочные последствия инфекции для здоровья человека неизвестны, однако мы все же можем предположить некоторые потенциальные эффекты воздействия вируса на продолжительность жизни клеток и здоровье организма в целом. В этой статье мы обсуждаем возможность того, что инфицирование SARS-CoV-2 в долгосрочной перспективе ускоряет старение клеток у выживших не только в пораженных тканях, но и в других тканях, в том числе в мозге. Поскольку некоторые последствия могут манифестировать через месяцы или годы после инфекции, необходимо будет пристально следить за состоянием пациентов, пострадавших от коронавирусной инфекции. Тщательное ведение документации может позволить нам установить вероятные взаимосвязи с заболеваниями, ассоциированные с пожилым возрастом, такими как болезнь Паркинсона и другие нейродегенеративные заболевания.
Воздействие SARS-CoV-2 на ключевые аспекты старения
Хотя исследования, детально раскрывающие молекулярные механизмы воздействия SARS-CoV-2 на клетки, еще не были проведены, недавние работы по картированию взаимодействий между вирусными белками и человеческим протеомом позволяют выявить возможные причинно-следственные связи с возраст-ассоциированными заболеваниями [10]. В этом исследовании были показаны взаимодействия вирусных белков с человеческими белками, которые вовлечены в каскады, связанные со старением, — транспорт везикул (Nsp6, Nsp7, Nsp10, Nsp13, Nsp15, Orf3a, E, Orf8), модификации липидов (S гликопротеин), процессинг и регуляцию РНК (Nsp8, N), обмен убиквитина (Orf10) и митохондриальную активность (Nsp4, Nsp8 и Orf9c).
Нуклеокапсидный белок (N) взаимодействует с маркером стрессовых гранул G3BP1, белком, противовирусная активность которого обусловлена индукцией врожденного иммунного ответа [11-13]. Такие взаимодействия могут ингибировать образование стрессовых гранул, тем самым манипулируя обменом РНК и синтезом белка клетки-хозяина [14]. Нуклеокапсид SARS-CoV-2 также связывается с LARP1 — репрессором трансляции mTOR [10]. Важно, что все таргетные белки экспрессируются как в легочной, так и в других тканях. В исследованиях, посвященных воздействию SARS-CoV и ВГ на судьбу клеток, часто наблюдаются изменения сигнальных путей, вовлеченных в клеточное старение [15], что поддерживает предположение о воздействии SARS-CoV-2 на эти пути (рис. 1).
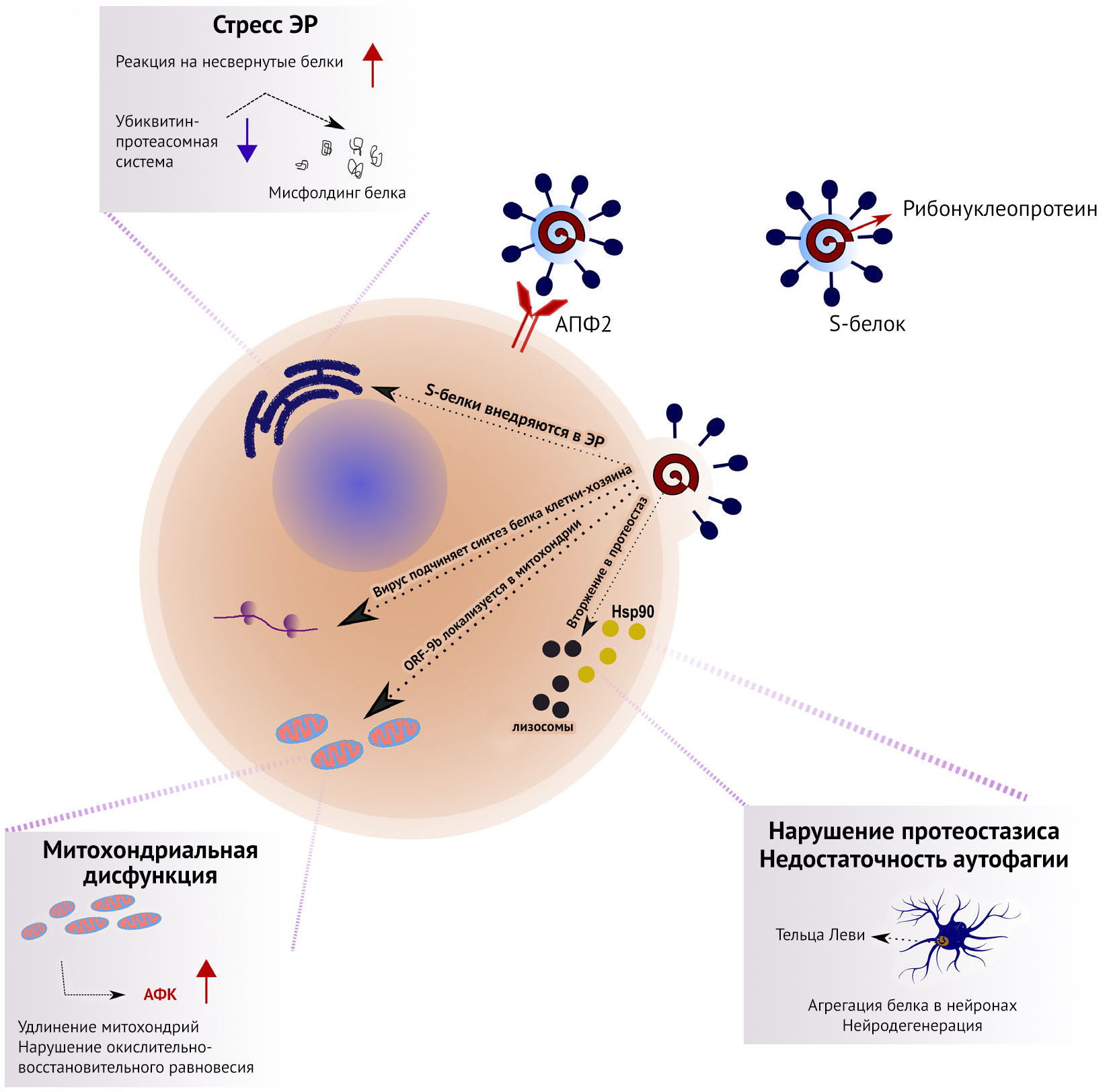
Протеостазис — это результат координированной работы множества структур, поддерживающих динамическое равновесие процессов трансляции белков, приобретения ими правильной конформации и их деградации. В нем участвуют молекулярные шапероны, в основном, белки теплового шока (Hsp), которые способствуют правильному сворачиванию белков, поддержанию их нативной конформации и участвуют в работе механизмов деградации. Однако поддержание стабильного протеома осложнено тем, что клетки часто подвергаются различным стрессам. Вирусная инфекция является одним из таких стрессоров, поскольку вирусы подчиняют себе молекулярные механизмы клетки-хозяина для нужд собственной репликации. В частности, вирусы используют несколько стратегий влияния на протеостазис на различных стадиях своего жизненного цикла. На ранних стадиях инфицирования H1N1 Hsp40 связывается с двумя субъединицами вирусной РНК полимеразы, повышая их активность [16]. Hsp40 способствует транслокации вирусного генома в ядро, которую связывают с взаимодействием с вирусными нуклеопротеидами (НП), которые инкапсулируют вирусный геном (рис. 1).
Более того, взаимодействие Hsp40 с НП играет роль на поздних стадиях инфицирования, ингибируя активацию протеинкиназы R (ПК-R), необходимой для антивирусного ответа хозяина [17,18]. Хотя предыдущие исследования показали, что при инфицировании H3N2 Hsp70 предотвращает экспорт рибонуклеопротеидов из ядра [19], новые исследования свидетельствуют о том, что Hsp70 действует как шаперон для вирусной полимеразы [20]. Еще одной целью ВГ служит Hsp90. После инфицирования Hsp90 перемещается в ядро и положительно регулирует активность и поддерживает структуру вирусных РНК-полимераз [21,22]. Белки теплового шока также являются негативными регуляторами клеточной смерти. В норме Hsp70, связываясь с Apaf-1 (индуктором апоптоза — прим. пер.), предотвращает включение прокаспазы-9 в апоптосому. Используя похожие механизмы, Hsp90 ингибирует олигомеризацию Apaf-1 и рекрутирование прокаспаз. Оба пути блокируют инициацию апоптоза [23,24]. При инфицировании вирусы могут предотвращать образование этих комплексов, облегчая активацию запускающих апоптоз каспаз для распространения инфекции и избегания иммунной системы хозяина.
Более того, SARS-CoV использует эндоплазматический ретикулум (ЭР) как место синтеза и процессинга вирусных белков [25]. Инфицирование SARS-CoV запускает реакцию на несвернутые белки (unfolded protein response — ЭР стресс, обусловленный накоплением необработанных шаперонами белков, вызывает повышение синтеза шаперонов и подавление синтеза секретируемых белков — прим. пер.). S-белки SARS-CoV активируют транскрипцию нескольких эффекторов UPR, таких как глюкоза-регулируемый белок 78 (GRP78), GRP94 и C/EBP-гомологичный белок. S-белки накапливаются в ЭР и, вероятно, напрямую влияют на UPR для облегчения вирусной репликации [25] (рис. 1).
Возможность деградации и утилизации внутриклеточных компонентов служит мощным инструментом уничтожения внутриклеточных патогенов [26]. Поэтому аутофагия, в ходе которой вирусы и вирусные белки переносятся в лизосомы для деградации, играет роль врожденной защиты против вирусов. Вирусы могут нарушать пути деградации белка для поддержания необходимой концентрации и функционирования вирусных белков. H1N1 блокирует аутофагию на ранних стадиях и приводит к снижению количества аутофагосом, а на поздних стадиях он подавляет слияние аутофагосом с лизосомами [27]. Однако аутофагосомы могут облегчать сборку репликаз РНК-вирусов с положительной цепью. Такой механизм был показан для неструктурированного белка (NSP) 6 птичьего коронавируса (птичий вирус инфекционного бронхита), который генерирует аутофагосомы из ЭР клетки-хозяина [28] (рис. 1). NSP6 ограничивает увеличение аутофагосом и тем самым способствует коронавирусной инфекции, препятствуя доставке вирусных компонентов в лизосомы для деградации. Открытая рамка считывания (ORF) 9b SARS-CoV запускает аутофагию клеток хозяина [29].
H1N1 может подчинять убиквитин-протеасомную систему хозяина. Клетки могут убиквитинировать вирусные белки, маркируя их для последующей деградации, однако у вирусов есть механизмы, позволяющие избежать деградации путем ингибирования клеточных антагонистов репликации вируса [30,31]. Эти механизмы вызывают нарушения протеостазиса, которые могут привести к накоплению токсичных нерастворимых белков [32]. В ответ на этот стресс клетки подавляют трансляцию генов домашнего хозяйства с целью сохранения энергии для синтеза стрессовых белков.
Известно, что SARS-CoV может влиять на митохондрии и их функционирование для избежания врожденного иммунного ответа [29]. ORF-9b SARS-CoV находится в митохондриях и обусловливает удлинение митохондрий путем повышения протеосомальной деградации динамин-подобного белка 1, который отвечает за деление митохондрий (рис. 1). Более того, ORF-9b воздействует на ассоциированную с митохондриями адаптерную молекулу MAVS, подавляя антивирусный клеточный сигналинг. Белки SARS-CoV ORF-3a, ORF-3b, ORF-6 и ORF-7a индуцируют апоптоз клетки-хозяина [33]. Другие адаптивные механизмы ответа на стресс — это секвестрация неправильно свернутых белков в стрессовых гранулах. H1N1 может ингибировать трансляцию и образование стрессовых гранул через фосфорилирование эукариотического фактора инициации трансляции 2а (eIF2a). Учитывая, что репликация вирусов зависит от работы механизмов трансляции клетки хозяина, множество вирусов связываются с ПК-R, чтобы предотвратить фосфорилирование eIF2a [34]. Многие внутриклеточные нарушения вызывают окислительно-восстановительный дисбаланс, повышая уровень активных форм кислорода (АФК), а также митохондриальную и лизосомальную дисфункцию. В итоге такая последовательность событий формирует порочный круг, делая клетки еще менее устойчивыми к инфицированию, что в долговременной перспективе может приводить к увеличению биологического возраста среди переживших коронавирусную инфекцию путем ускорения старения иммунной системы и пораженных тканей.
Возможные связи с болезнью Паркинсона
Нарушения протеостазиса, связанные с возрастом, коррелируют с наиболее тяжелыми последствиями ВГ и SARS-CoV-2 у пожилых. Потеря способности нормальной активации механизмов ответа на стресс у пожилых может приводить к серьезным фенотипам, в том числе к формированию малорастворимых белковых агрегатов, свойственных также и нейродегенеративным заболеваниям, таким как болезнь Паркинсона. Инфицирование H1N1 дофаминергических клеток, экспрессирующих альфа-синуклеин (aSyn), основной компонент телец Леви и нейритов Леви, приводит к формированию aSyn агрегатов, но не агрегатов тау или TDP-43 [27]. Выявленные в этом исследовании молекулярные механизмы указывали на блокирование аутофагии H1N1, что давно связывали с аккумуляцией aSyn в моделях болезни Паркинсона.
В этом контексте интересно, что антивирусный препарат амантадин используют на ранних стадиях болезни Паркинсона для терапии тремора [35–39]. Кроме того, оcельтамивир, противовирусный препарат, широко используемый в терапии гриппа, значительно снижает выраженность паркинсонизма, однако в то же время усиливает дискинезии [40]. Хотя не выявлено связи между инфицирования вирусом гриппа в прошлом и развитием идиопатической болезни Паркинсона, паркинсонизм может быть связан с перенесенными инфекциями [41].
aSyn может играть роль в активации врожденного и приобретенного иммунитета при болезни Паркинсона [42,43]. Таким образом, исследования молекулярных механизмов взаимосвязи вирусных инфекций с нарушением клеточного протеостазиса, которые может усилить агрегацию aSyn, может привнести новые терапевтические стратегии в терапию болезни Паркинсона и повлиять на терапию пациентов, страдающих болезнью Паркинсона и перенесших инфицирование SARS-CoV-2.
В ранних исследованиях были показаны возможные взаимодействия человеческого коронавируса с ЦНС и болезнью Паркинсона в том числе [44]. После внутримозговых инъекций ВГ у мышей вирус обнаруживался в черной субстанции и в гиппокампе [45].
Важным является и то, что aSyn может служить противовирусным фактором у пациентов с энцефалитом Западного Нила. У мышей, нокаутных по aSyn, была значительно повышена смертность от вируса лихорадки Западного Нила, и титр вируса в ЦНС был на пять порядков больше, чем у мышей дикого типа. Активация вирус-индуцированной каспазы-3 в нейронах коры нокаутных по aSyn мышей наблюдалась раньше, что гораздо раньше приводило к апоптозу нейронов [46].
Мутации в обогащенной лейциновыми повторами киназе (LRRK2) являются самой распространенной известной генетической детерминантой болезни Паркинсона. У мышей, экспрессирующих мутантную LRRK2G2019S, была повышена смертность от реовирус-индуцированного энцефалита. В головном мозге этих животных обнаружили повышенную концентрацию aSyn [47].
LRRK2 экспрессируется во многих типах иммунных клеток, и его экспрессия повышается при стимуляции макрофагов патогенами [48]. Предыдущие исследования предоставляют достаточно данных, чтобы предположить, что LRRK2 в иммунной системе связывает иммунные процессы и развитие и распространение болезни Паркинсона, а также биологическое старение клеток [48].
