
Взаимодействие лекарственных веществ и их мишеней

По приблизительным оценкам лекарственные препараты, постоянно используемые в клинической практике, имеют свыше 500 целевых структур в человеческом организме, воздействуя на которые лекарственные соединения реализуют свой терапевтический эффект. Но каким бы ни было число мишеней, количественно они все равно уступают многообразию белков, задействованных в биохимических процессах организма. Несмотря на то, что по мере изучения белков, принимающих участие в развитии патологических состояний, их стараются использовать в качестве очередной мишени для терапевтического воздействия, путь от определения гена, ответственного за синтез того или иного белка, до понимания функции белка и его роли в клеточных и организменных процессах долог и тернист.
Для многих белков, кодируемых в человеческом геноме, определено их отношение к тому или иному семейству, что возможно благодаря сравнительному анализу последовательностей: извлекается кодирующая геномная последовательность, определяется транслируемая часть гена, составляется последовательность белкового продукта, производится поиск сходных для этой последовательности известных белков.
Однако как выглядит пространственная структура белка, для которого выявлена какая-либо последовательность аминокислот? Какие лиганды белок способен распознавать и связывать и какой биохимический толк в его работе?
Ответы на эти вопросы уже не получить без серии серьезных экспериментов. Но для начала немного разберемся вот в чем: хотелось бы сразу определить, что под биохимической функцией белка подразумевается по сути то, чем является это белок – протеаза, ионный канал, переносчик или что угодно другое, но данная информация о белке, увы, не даст нам сразу представления и о системных задачах белка, которые он выполняет в пределах клетки или же целого организма. За это ответственна пространственная структура белка, именно потому к ее изучению приковано столько внимания ученых. Уже на сегодняшний день для многих семейств генов удалось определить структуру всех их членов, а вот расшифровка пространственной организации соответствующих белков требует существенных затрат и потому запаздывает.
Также хотелось бы обратить ваше внимание на основные обобщенные концепты для разработки любого нового лекарственного вещества: то есть, будет ли данное соединение ингибитором, агонистом, антагонистом или же аллостерическим регулятором. Как будет выглядеть любой из представителей данных групп, определяется структурой мишени, на которую данное соединение будет направлено.
Прежде, чем рассмотреть взаимодействие лекарственных соединений с их мишенями, вспомним, каким образом происходит подготовка субстратов в организме. Ни один метаболический путь, синтез или регуляторный процесс не обходится без катализаторов.
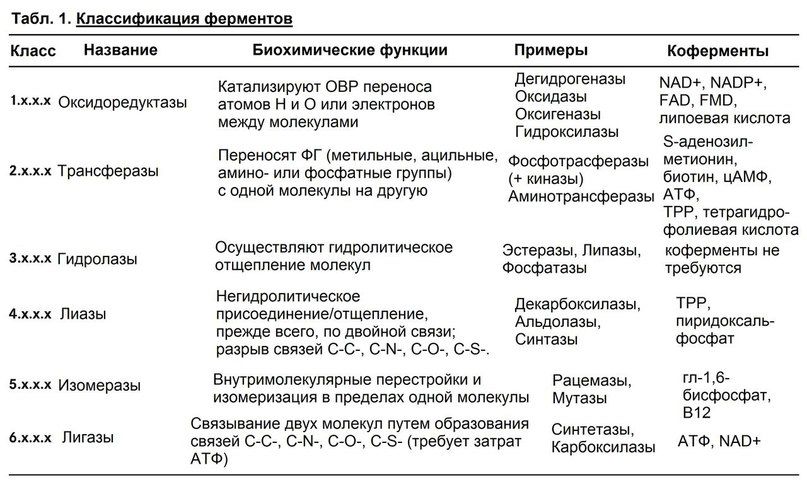
Табл. 1. Классификация ферментов. Четырехзначный шифр для каждого фермента подразумевает его класс, подкласс, подподкласс и индивидуальный номер фермента.
Ферменты связываются со своими субстратами и продуктами превращения не прочно. Конформация лиганда в связанном состоянии нередко отличается от конформации, которая была бы энергетически наиболее выгодна в водном растворе. Фермент связывает субстрат согласно той геометрии, которая позволит субстрату принять форму переходного состояния, подразумеваемого осуществляемой реакцией.
Кроме того, полярные группы могут индуцировать смещения зарядов, что тоже следует учитывать. Посредством структуры и расположения активных групп фермент стабилизирует соединение переходного состояния (хотя, замечу, что переходное состояние не является химическим соединением в обычном смысле слова, т.е. к нему не применимы понятия и закономерности, связанные с настоящими химическими соединениями (валентность, постоянство атомных расстояний и др.). В то же время, благодаря ферментам, происходит снижение энергии активации, вследствие чего ход реакции существенно ускоряется. После получения продукта фермент всегда снова готов преобразовать следующую молекулу субстрата.
Чтобы лучше разобраться в том, как же фермент готовит субстрат к переходному состоянию, возьмем небольшой пример.
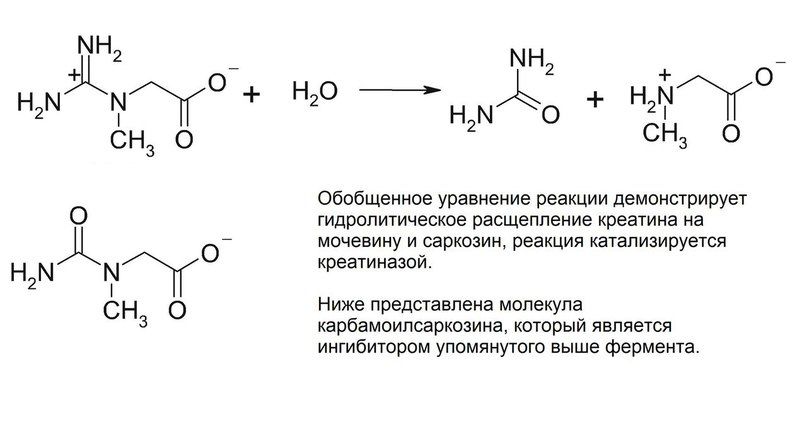
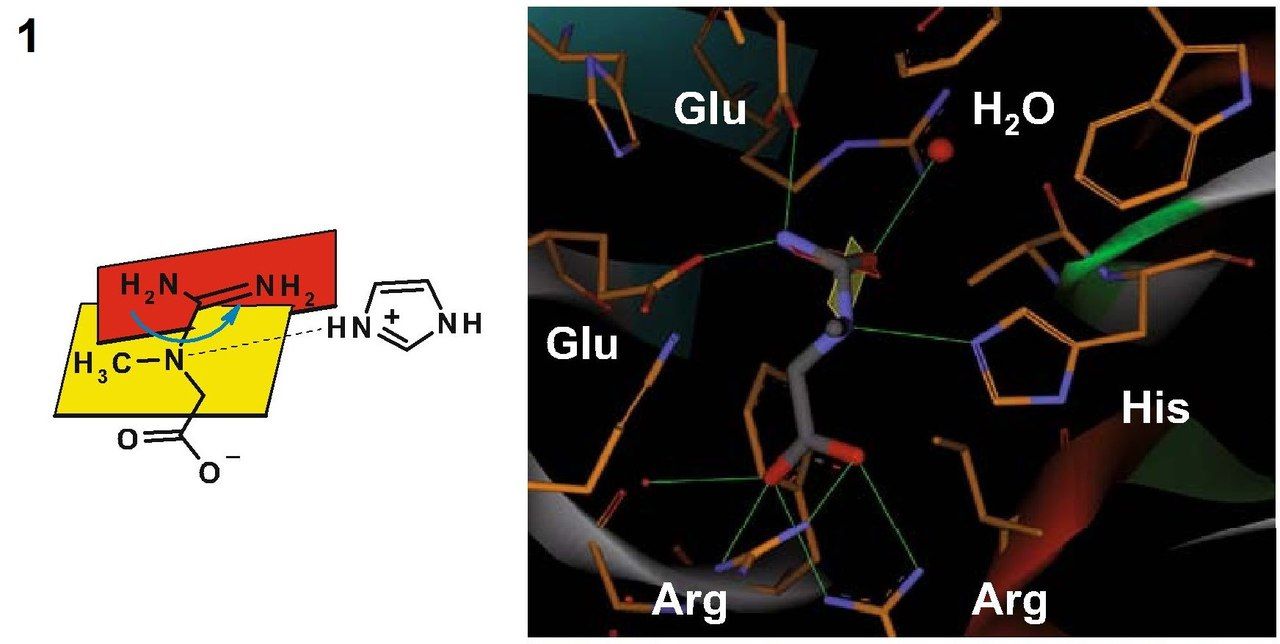
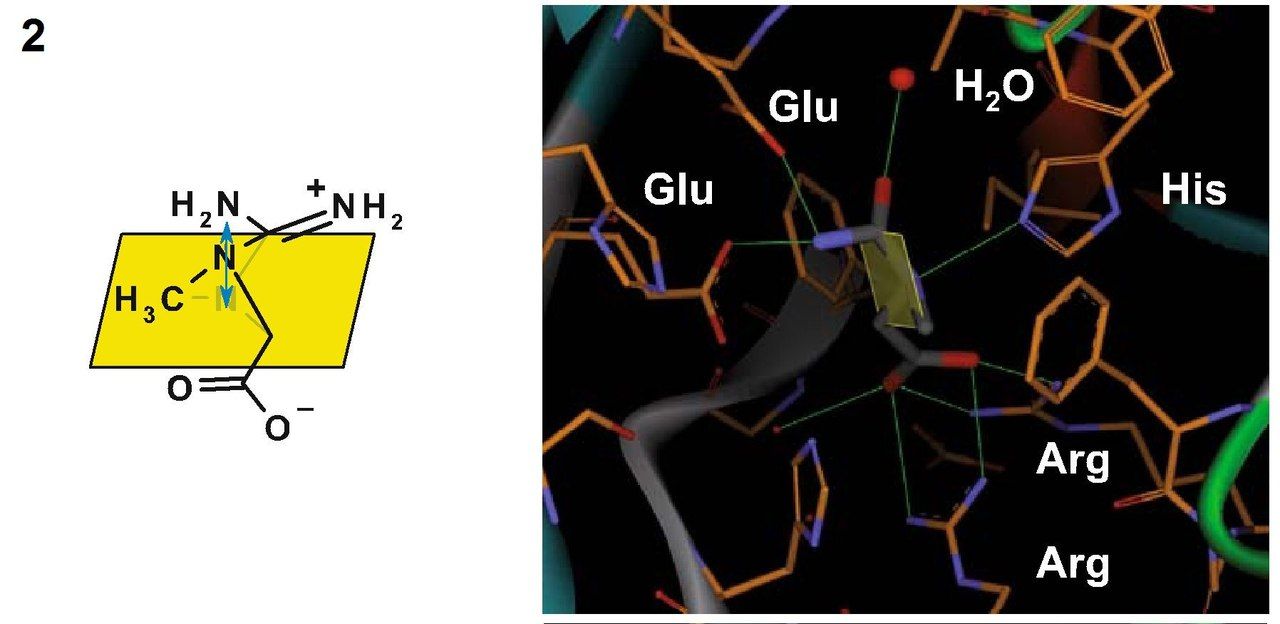
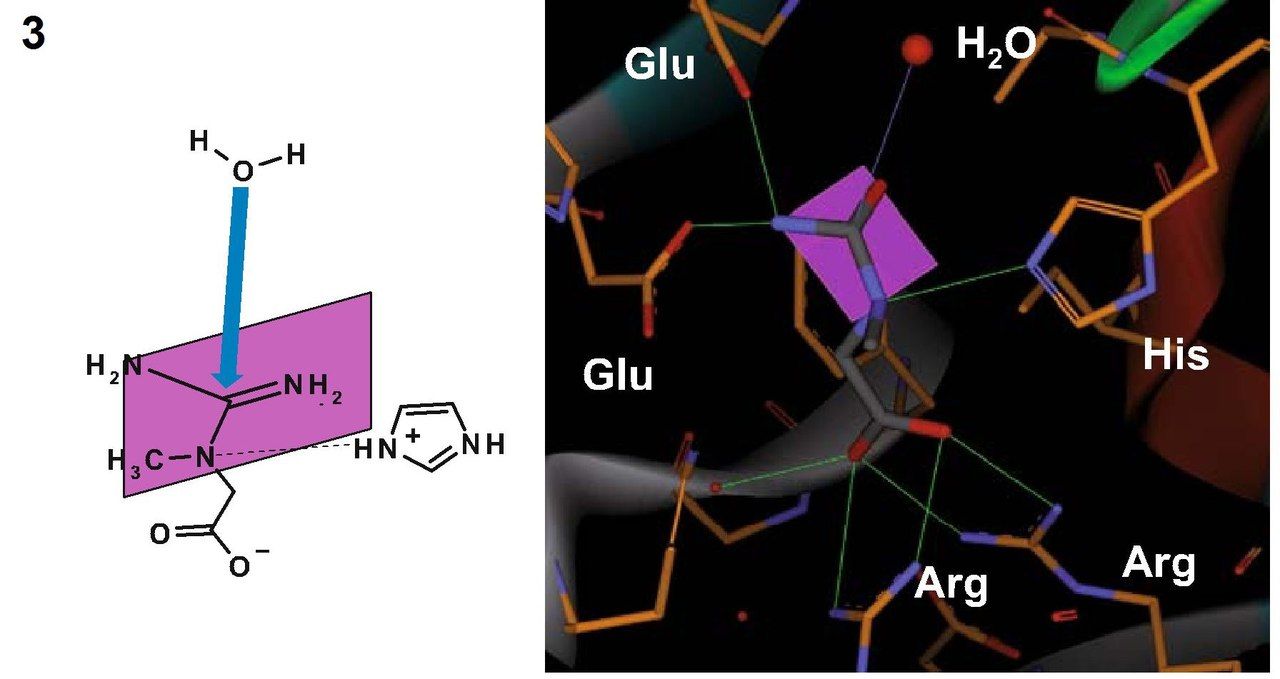
Кристаллическая структура креатиназы была определена с помощью карбамоилсаркозина, близкому по структуре к естественному субстрату данного фермента – креатину. Фермент катализирует расщепление креатина до мочевины и саркозина. Для осуществления этой реакции должен произойти разрыв связи C-N в гуанидиновой группе креатина путем нуклеофильного присоединения молекулы воды к центральному атому углерода. Вследствие делокализации электронов все три C-N связи в гуанидиновой группе приобретают частичные двойные связи, и молекула становится плоской. Как раз теперь наступает момент исполнения ферментом своей задачи – ему необходимо изменить пространственную геометрию креатина, чтобы сделать возможным переходное состояние и последующее нуклеофильное присоединение. Креатин, как цвиттер-ионное соединение, связывается двумя остатками глутамата посредством образования водородных мостиков между гуанидиновой группой и этими самыми остатками глутамата, а находящаяся на противоположном конце молекулы кислотная группа образует с двумя остатками аргинина сильно поляризованные связи. Теперь обратим внимание на молекулу воды, находящуюся вблизи центрального атома С гуанидиновой группы. В кармане для связывания у нашего фермента имеется гистидин. И этот гистидин не только позволяет молекуле воды оказаться в нужном расположении, но и отщепляет от нее протон, поскольку оставшаяся гидроксогруппа обладает высокой нуклеофильностью. Фиксация гуанидиновой группы остатками глутамата делает возможным вращение не связанных планарных частей молекулы креатина, вследствие чего конъюгированные связи разрываются и связь C-N, по которой впоследствии и произойдет расщепление, существенно ослабляется. Нуклеофильное присоединение становится возможным и формируется переходное состояние. В то же время протонированный гистидин (помните, отщепили от воды протон?) поляризует связанный с метильной группой атом азота в молекуле креатина и связывается с ним водородной связью. Тем самым фермент готовит молекулу к переходному состоянию, соответствующему разрыву связи. После переноса протона с гистидина на субстрат атом азота в разрываемой связи приобретает положительный заряд. Гистидин забирает себе протон от кислорода, который входит теперь в состав тетраэдрического комплекса. Одновременно с этими событиями формируется двойная связь между С и О, а центральная C-N связь разрывается. На этом моменте продукты покидают карман связывания фермента.
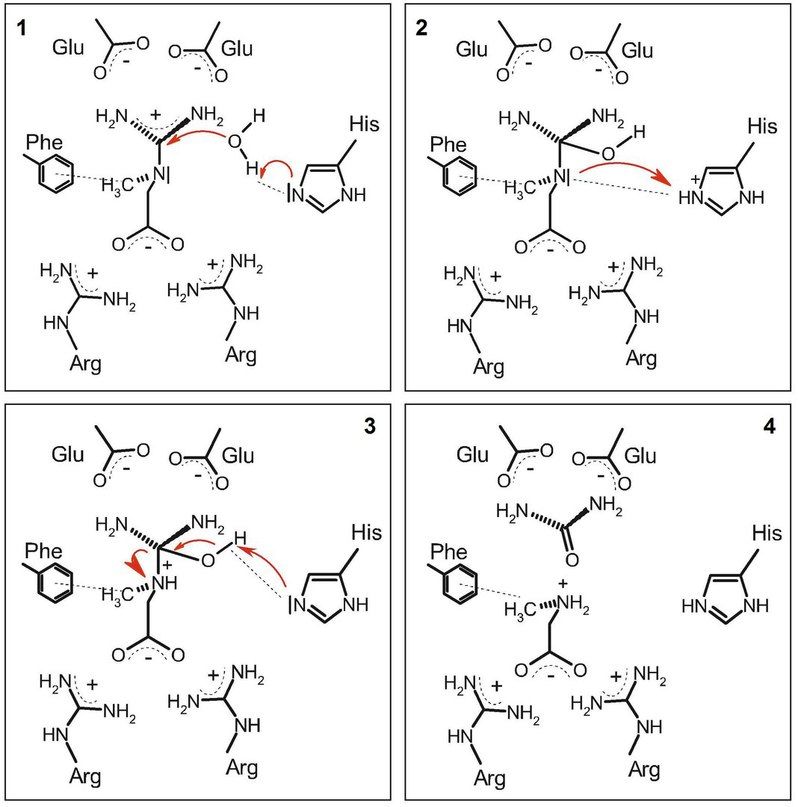
1 — Гистидин поляризует молекулу воды, что опосредует последующую нуклеофильную атаку по центральному атому С гуанидиновой группы.
2 — Гистидин переносит протон на центральный атом азота.
3 — Образование двойной связи между атомами С и О; разрыв связи C-N.
4 — Продукты реакции - мочевина и саркозин - покидают карман связывания фермента.
Таким образом, фермент создает требуемое стерическое и электронное окружение для осуществляемой реакции. Полярные остатки, содержащиеся в ферменте, позволяют правильно расположить молекулу воды для нуклеофильного присоединения, а гистидин обуславливает пирамидальное строение и положение атома азота в разрываемой связи. Помимо этого, фермент также служит как донором, так и акцептором протонов.
Как уже было упомянуто выше, кристаллическая структура фермента была определена с помощью не самого креатина, а сходного с ним соединения – карбамоилсаркозина. Его молекула отличается от креатина лишь заменой одного атома кислорода на атом азота. Однако по этой причине данная часть молекулы уже не будет заряжена положительно, как это происходит с креатином. Нуклеофильное присоединение гидроксогруппы провоцирует в молекуле креатина разрыв связи в гуанидиновой группе и выравнивание зарядов. Но в молекуле карбамоилсаркозина подобное присоединение привело бы к возникновению отрицательного заряда, что в непосредственной близости к имеющимся отрицательно заряженным глутаматам энергетически крайне не выгодно. Соответственно, далее никакого расщепления не происходит и реакция попросту блокируется. Это наглядный пример того, насколько фермент и его субстрат должны быть структурно и электронно согласованы. Малейшее изменение – и вот уже молекула субстрата превратилась в ингибитор и превращения, которое стремится осуществить фермент, не происходит.
Помимо одиночной работы, ферменты могут организовываться в мультиферментные комплексы, которые могут проводить над одним субстратом цепочку последовательных превращений. Между собой такие комплексы ферментов могут образовывать целые каскадные системы, в которых каждый предыдущий фермент активирует соединения для дальнейшего преобразования последующим ферментом. К примеру, в рамках каскада свертывания крови активация происходит двумя независимыми путями, каждый из которых включает в себя несколько этапов. В конечном итоге оба пути сводятся к одному процессу. Таким образом, даже слабое пусковое воздействие может быть многократно усилено с помощью каскадно протекающих реакций. В случае запуска свертывающего каскада при каких-либо повреждениях, данное усиление процесса играет положительную роль. Но, к примеру, если повышенная свертываемость крови обусловлена обстоятельствами какого-то заболевания, то последствия могут быть уже отрицательными. Для таких случаев, когда необходимо предотвратить действие фермента, существуют различные способы их ингибирования.
Ингибиторы могут предотвратить каталитическое действие фермента, заняв место, предназначенное для связывания субстрата. Такие ингибиторы будут называться конкурентными. Кроме них есть аллостерические ингибиторы, которые, связываясь с ферментом, изменяют его трехмерную структуру или динамические свойства, что может препятствовать ферменту принять необходимую для катализа конформацию и вести к снижению его активности. Рассматривая детально ферментативную кинетику, можно дополнительно выделить конкурентное и неконкурентное ингибирование. Также по типу взаимодействия с ферментом различают ингибиторы обратимого и необратимого действия.
Обратимые ингибиторы должны обладать высоким сродством к ферменту, чтобы надежно предотвратить превращение субстрата. Некоторые обратимые ингибиторы ковалентно связываются с каталитическим центром, однако такая связь химически лабильна и потому связывание полностью обратимо (например, полуацетальная связь).
Необратимые же ингибиторы на то и необратимые, чтобы их связь с ферментом нельзя было разорвать, и потому фермент остается неактивным вплоть до его разрушения в организме. Также есть протеазные ингибиторы, которые хотя и связываются обратимо, но связь довольно прочная, и потому комплекс ингибитора и фермента успевает разрушиться, прежде чем ингибитор отсоединится. Добавлю, что необратимые ингибиторы находятся в меньшинстве по сравнению с обратимыми, тем не менее, немало значимых препаратов относятся именно к данной группе (например, ацетилсалициловая кислота, омепразол, клопидогрел, пенициллины, цефалоспорины, ингибиторы МАО). Итак, рациональный дизайн ферментных ингибиторов основывается, преимущественно, на структуре субстрата конкретного фермента. Но это уже совсем другая история!
Источники:
- Breitmaier E., Jung G. Organische Chemie: Grundlagen, Stoffklassen, Reaktionen, Konzepte, Molekülstrukturen; 129 Tabellen. – Georg Thieme Verlag, 2012.
- Imming P., Sinning C., Meyer A. Drugs, their targets and the nature and number of drug targets //Nature reviews Drug discovery. – 2006. – Т. 5. – №. 10. – С. 821.
- Brückner R. Reaktionsmechanismen, 3. Auflage, 2004.
