
Ингибирование Chk1: двойной удар по плоскоклеточному раку

Плоскоклеточный рак органов головы и шеи (ПКР ОГШ) развивается в слизистой оболочке верхних отделов пищеварительного тракта и ежегодно составляет ~ 700 000 (5 %) впервые диагностированных случаев рака во всем мире, при этом 5-летняя выживаемость пациентов остается невысокой и составляет около 60 %. Курение, употребление алкоголя и инфицирование высокоонкогенными типами вируса папилломы человека являются известными факторами риска этого заболевания [1].
Стандартизированные протоколы лечения включают хирургическую резекцию опухоли, лучевую терапию и химиотерапию препаратами платины, часто в сочетании, что приводит к тяжелым побочным эффектам. Единственной целевой терапией, одобренной для ПКР ОГШ, является цетуксимаб — химеризованное моноклональное антитело рецептора эпидермального фактора роста (EGFR) [2].
Исследователями Атласа ракового генома (The Cancer Genome Atlas, TCGA) опубликован подробный молекулярный ландшафт соматических мутаций ПКР ОГШ. Отсутствие мутаций драйверных онкогенов затрудняет идентификацию новых терапевтических мишеней, но обнаружено большое количество мутаций в генах-супрессорах опухолей, ассоциированных с клеточным циклом. В подавляющем большинстве образцов ПКР ОГШ обнаружена аберрантная экспрессия TP53, возникшая в результате мутаций или инактивации онкопротеином ВПЧ Е6. Кроме того, утрачивается функция CDKN2A/p16, а циклин D1 часто сверхэкспрессируется, что в совокупности приводит к нарушению контрольной точки G1/S и далее G2/M. Потеря регуляции G1/S-перехода вызывает незапланированное вступление клетки в S-фазу и стресс репликации, зачастую приводящий к повреждению ДНК, что придает белкам, контролирующим S-фазу и переход G2/M, ключевую роль. Наиболее вероятными кандидатами на роль мишеней для лечения этого типа рака рассматривают участников системы контроля повреждений ДНК: протеинкиназы ATM (мутантный белок при атаксии-телеангиэктазии) и ATR (АТМ- и Rad3-родственная киназа), а также серин/треонин-киназы Chk1 и Chk2 с функцией чекпойнтов [3].
Anne M. van Harten с коллегами исследовали канонический путь ATM-CHEK2/ATR-CHEK1 в качестве специфической мишени ПКР ОГШ. Интерференция или ингибирование РНК ATM, ATR и CHEK2 не вызывали гибели опухолевых клеток, при этом Chk1-ассоциированная передача сигналов критична для успешного прохождения опухолевыми клетками S-фазы клеточного цикла (Рис.1) [4]. Блокирование функции Chk1 (специфическим ингибитором LY2606368 / Prexasertib) приводило к прекращению репликации ДНК, «коллапсу» вилки репликации и накоплению повреждений ДНК. Гибель клеток в данном случае происходит по бимодальному механизму. В наиболее чувствительных клеточных линиях апоптоз индуцируется в S-фазе, тогда как более резистентные клеточные линии способны избегать апоптоза в репликативной фазе, но в них накапливаются хромосомные аберрации, которые становятся летальными в последующем митозе. Кроме того, чувствительность к ингибированию Chk1 требует наличия функционирующих циклинзависимых киназ CDK1 и CDK4/6. То есть ингибирование Chk1 не следует сочетать с ингибиторами CDK1 (например, RO-3306) или CDK4/6 (Палбоциклиб, применяемый для терапии HR-положительного и HER2-негативного рака молочной железы) или другими лекарственными средствами, которые препятствуют прогрессированию клеточного цикла, поскольку является необходимым условием для эффективного ингибирования Chk1. И напротив, его комбинация с ингибиторами, стимулирующими дальнейшее прогрессирование клеточного цикла, оказывает аддитивное действие. Уровень экспрессии CDK1 может использоваться в качестве потенциального критерия отбора пациентов и предикции ответа на терапию ингибиторами Chk1 (обратная корреляция с реакцией ингибирования) [5].
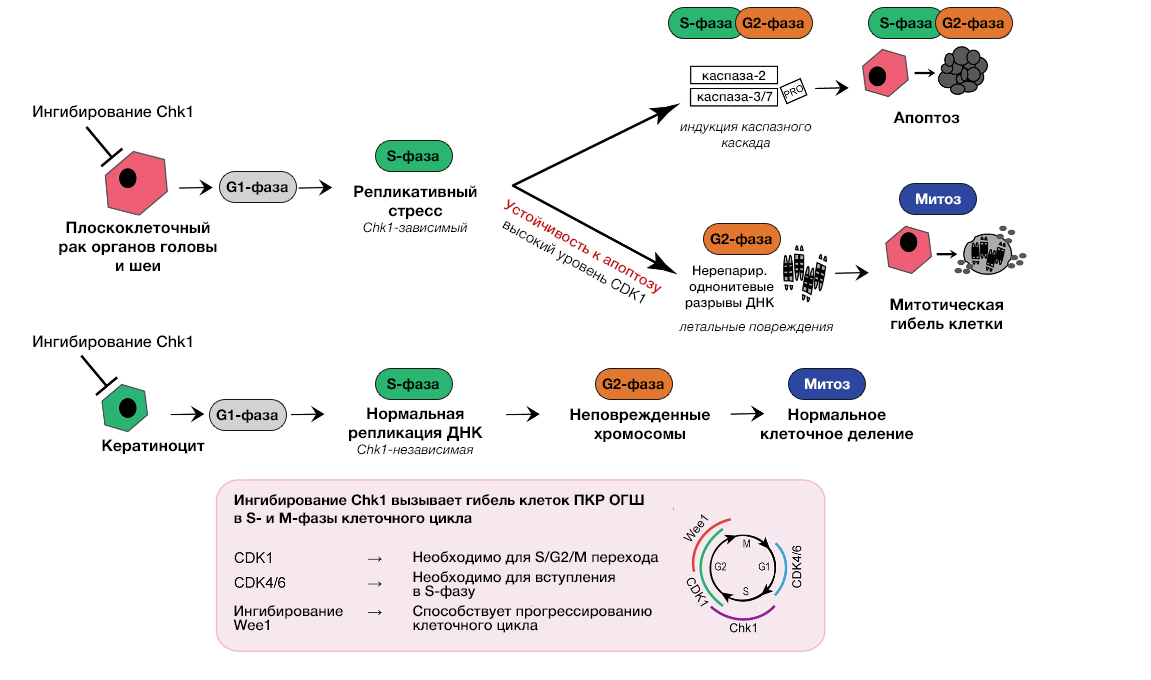
Рисунок 1 | Схема молекулярного механизма чувствительности опухолевых клеток ПКР ОГШ.
При ингибировании Chk1 клетки «застревают» в S-фазе клеточного цикла из-за нарушения репликации ДНК, что приводит к накоплению ее повреждений. В S- или ранней G2-фазе инициируется каспазный каскад с последующей апоптотической гибелью клеток с низким собственным уровнем CDK1 (чувствительные клетки). Клетки с высоким CDK1 сталкиваются с аналогичными проблемами репликации ДНК, но не инициируют апоптоз и переходят далее в митоз с неполной репликацией ДНК и сохраненными нерепарированными повреждениями ДНК. Это приводит к хромосомным аберрациям, вызывающим гибель во время последующего деления клетки. Эффективность ингибирования Chk1 зависит от прогрессирования клеточного цикла. Следовательно, функционально активный CDK4/6 необходим для входа в S-фазу, CDK1 — для прогрессирования в S-фазе и последующего входа в митоз, что тем самым способствует накоплению повреждений ДНК. Комбинация ингибиторов Chk1 с препаратами, тормозящими клеточный цикл, неэффективна; напротив, поддержка перехода в митоз способствует гибели более резистентных агрессивных опухолевых пулов клеток.
Обсуждается потенциальная противоопухолевая сенсибилизирующая активность препарата адавосертиб, который избирательно ингибирует Wее1 — тирозинкиназу, фосфорилирующую CDK1 для последующей инактивации комплекса CDK2 / циклин B. Ингибирование активности Wee1 предотвращает фосфорилирование CDR2 и блокирует контрольную точку G2, что может привести к апоптозу при лечении химиотерапевтическими агентами, повреждающими ДНК. В отличие от нормальных, в большинстве злокачественно трансформированных клеток с дефицитом или мутацией p53 отсутствует контрольная точка G1, поскольку p53 является ее ключевым регулятором, и в этих клетках ключевой становится контрольная точка G2 для восстановления ДНК в поврежденных клетках. Поэтому блок G2 делает опухолевые клетки с дефицитом p53 более уязвимыми для противоопухолевых агентов и усиливает их цитотоксический эффект [6].
Источники:
- Suh, Y. et al. Clinical update on cancer: molecular oncology of head and neck cancer. Cell death and disease 5.1 (2014): e1018.
- Ling, D. C., Bakkenist, C. J., Ferris, R. L., Clump, D. A. Role of immunotherapy in head and neck cancer. Semin. Radiat. Oncol.28, 12–16 (2018).
- Leemans, C. R., Snijders, P. J. F. , Brakenhoff, R. H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18, 269–282 (2018).
- Anne M. van Harten et al.Targeting the cell cycle in head and neck cancer by Chk1 inhibition: a novel concept of bimodal cell death. Oncogenesis. 8 (38) (2019).
- Ansari S.S. et al. Upregulation of cell cycle genes in head and neck cancer patients may be antagonized by erufosine's down regulation of cell cycle processes in OSCC cells. Oncotarget. 2017;9(5):5797-5810.
- Lawrence, M. S. et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015).
